

Definition of functionally and structurally distinct repressive states in the nuclear receptor PPARγ
- PMID: 31862968
- PMCID: PMC6925260
- DOI: 10.1038/s41467-019-13768-0
Definition of functionally and structurally distinct repressive states in the nuclear receptor PPARγ
Abstract
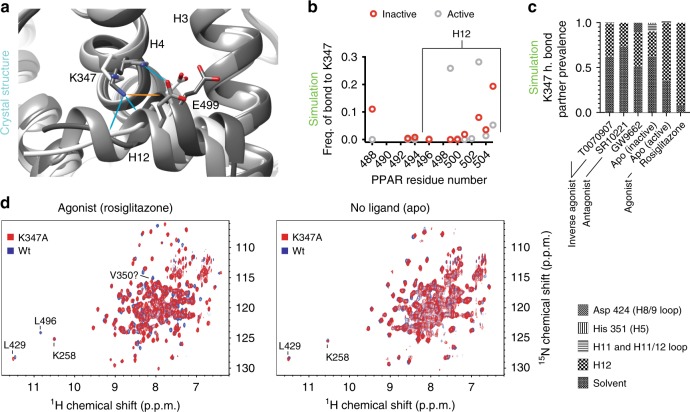
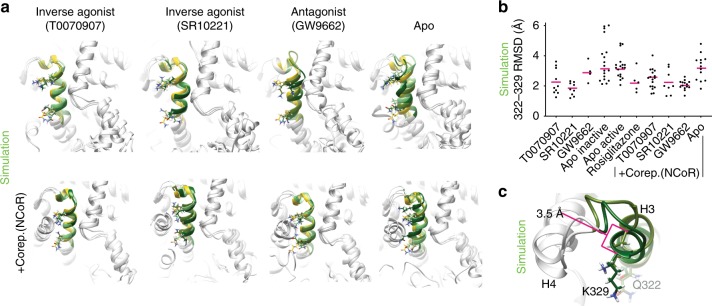
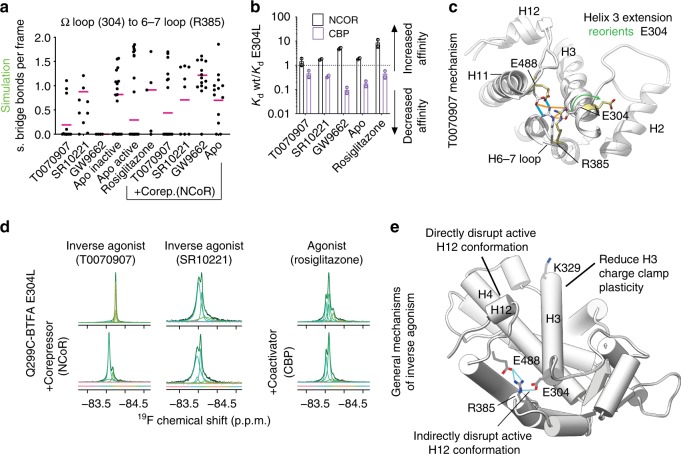
The repressive states of nuclear receptors (i.e., apo or bound to antagonists or inverse agonists) are poorly defined, despite the fact that nuclear receptors are a major drug target. Most ligand bound structures of nuclear receptors, including peroxisome proliferator-activated receptor γ (PPARγ), are similar to the apo structure. Here we use NMR, accelerated molecular dynamics and hydrogen-deuterium exchange mass spectrometry to define the PPARγ structural ensemble. We find that the helix 3 charge clamp positioning varies widely in apo and is stabilized by efficacious ligand binding. We also reveal a previously undescribed mechanism for inverse agonism involving an omega loop to helix switch which induces disruption of a tripartite salt-bridge network. We demonstrate that ligand binding can induce multiple structurally distinct repressive states. One state recruits peptides from two different corepressors, while another recruits just one, providing structural evidence of ligand bias in a nuclear receptor.
Conflict of interest statement
The authors declare no competing interests.
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
- P20 GM103546/GM/NIGMS NIH HHS/United States
- R00 DK103116/DK/NIDDK NIH HHS/United States
- R00DK103116/U.S. Department of Health & Human Services | NIH | National Institute of Diabetes and Digestive and Kidney Diseases (National Institute of Diabetes & Digestive & Kidney Diseases)/International
- P20GM103546/U.S. Department of Health & Human Services | NIH | National Institute of General Medical Sciences (NIGMS)/International
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
