

Patterns of diverse gene functions in genomic neighborhoods predict gene function and phenotype
- PMID: 31863070
- PMCID: PMC6925100
- DOI: 10.1038/s41598-019-55984-0
Patterns of diverse gene functions in genomic neighborhoods predict gene function and phenotype
Abstract
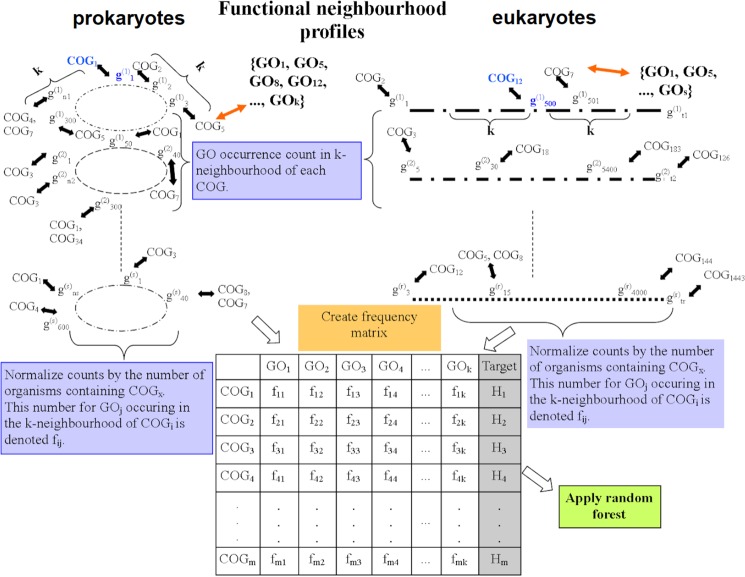
Genes with similar roles in the cell cluster on chromosomes, thus benefiting from coordinated regulation. This allows gene function to be inferred by transferring annotations from genomic neighbors, following the guilt-by-association principle. We performed a systematic search for co-occurrence of >1000 gene functions in genomic neighborhoods across 1669 prokaryotic, 49 fungal and 80 metazoan genomes, revealing prevalent patterns that cannot be explained by clustering of functionally similar genes. It is a very common occurrence that pairs of dissimilar gene functions - corresponding to semantically distant Gene Ontology terms - are significantly co-located on chromosomes. These neighborhood associations are often as conserved across genomes as the known associations between similar functions, suggesting selective benefits from clustering of certain diverse functions, which may conceivably play complementary roles in the cell. We propose a simple encoding of chromosomal gene order, the neighborhood function profiles (NFP), which draws on diverse gene clustering patterns to predict gene function and phenotype. NFPs yield a 26-46% increase in predictive power over state-of-the-art approaches that propagate function across neighborhoods, thus providing hundreds of novel, high-confidence gene function inferences per genome. Furthermore, we demonstrate that copy number-neutral structural variation that shapes gene function distribution across chromosomes can predict phenotype of individuals from their genome sequence.
Conflict of interest statement
The authors declare no competing interests.
Figures





References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
