

ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?
- PMID: 31866818
- PMCID: PMC6909825
- DOI: 10.3389/fnins.2019.01310
ALS Genetics, Mechanisms, and Therapeutics: Where Are We Now?
Abstract
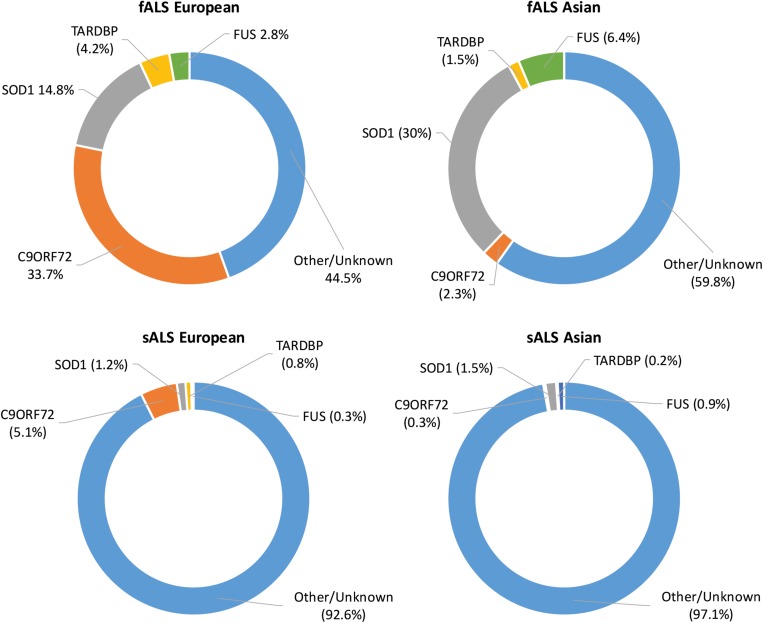
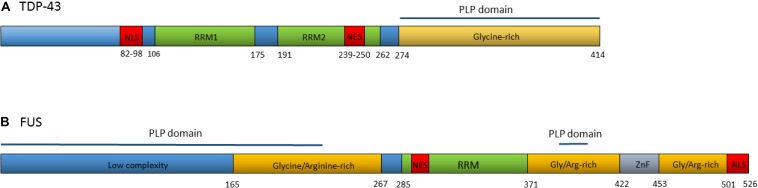
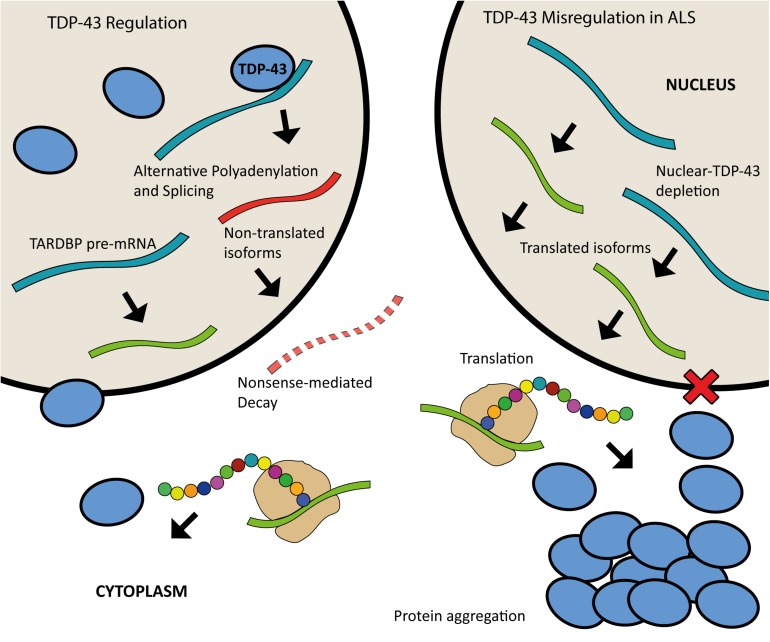
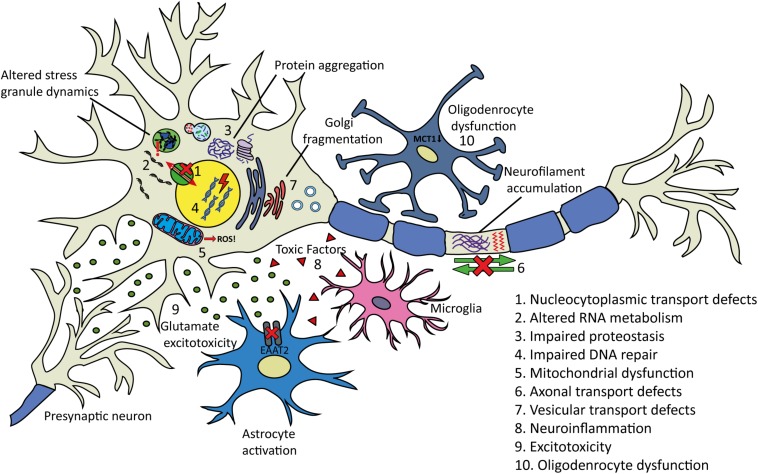
The scientific landscape surrounding amyotrophic lateral sclerosis (ALS) continues to shift as the number of genes associated with the disease risk and pathogenesis, and the cellular processes involved, continues to grow. Despite decades of intense research and over 50 potentially causative or disease-modifying genes identified, etiology remains unexplained and treatment options remain limited for the majority of ALS patients. Various factors have contributed to the slow progress in understanding and developing therapeutics for this disease. Here, we review the genetic basis of ALS, highlighting factors that have contributed to the elusiveness of genetic heritability. The most commonly mutated ALS-linked genes are reviewed with an emphasis on disease-causing mechanisms. The cellular processes involved in ALS pathogenesis are discussed, with evidence implicating their involvement in ALS summarized. Past and present therapeutic strategies and the benefits and limitations of the model systems available to ALS researchers are discussed with future directions for research that may lead to effective treatment strategies outlined.
Keywords: FUS; TDP-43; amyotrophic lateral sclerosis; cell models; disease mechanisms; missing heritability; therapeutics.
Copyright © 2019 Mejzini, Flynn, Pitout, Fletcher, Wilton and Akkari.
Figures
References
-
- Abe K., Itoyama Y., Sobue G., Tsuji S., Aoki M., Doyu M., et al. (2014). Confirmatory double-blind, parallel-group, placebo-controlled study of efficacy and safety of edaravone (MCI-186) in amyotrophic lateral sclerosis patients. Amyotroph. Lateral Scler. Frontotemporal Degener. 15 610–617. 10.3109/21678421.2014.959024 - DOI - PMC - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous