

Antimicrobial Peptide Designing and Optimization Employing Large-Scale Flexibility Analysis of Protein-Peptide Fragments
- PMID: 31867532
- PMCID: PMC6921640
- DOI: 10.1021/acsomega.9b03035
Antimicrobial Peptide Designing and Optimization Employing Large-Scale Flexibility Analysis of Protein-Peptide Fragments
Abstract
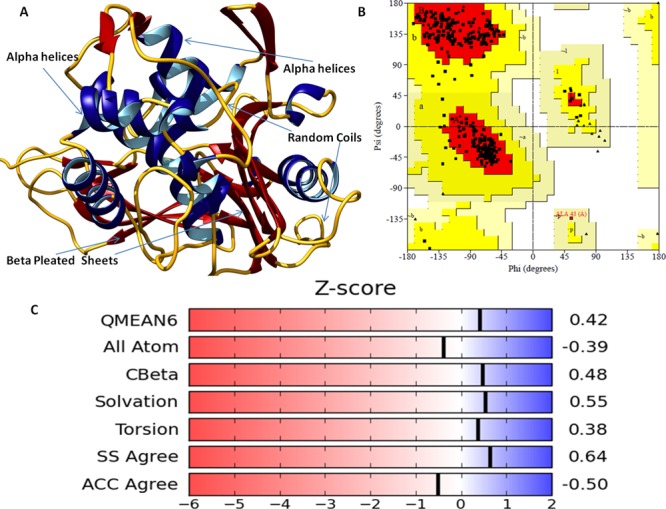
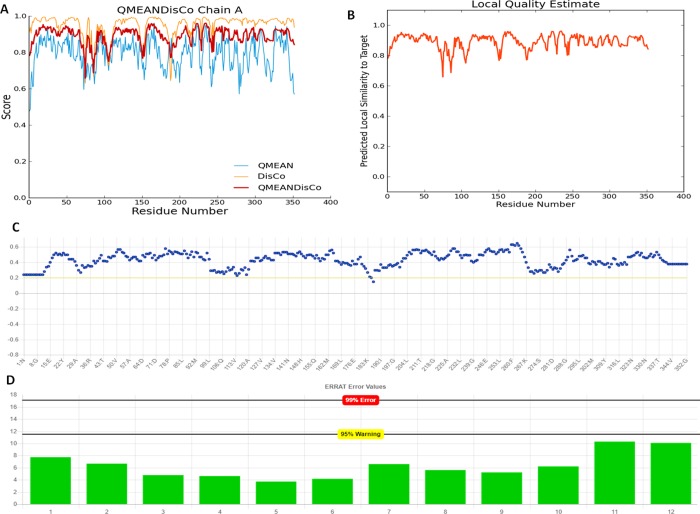
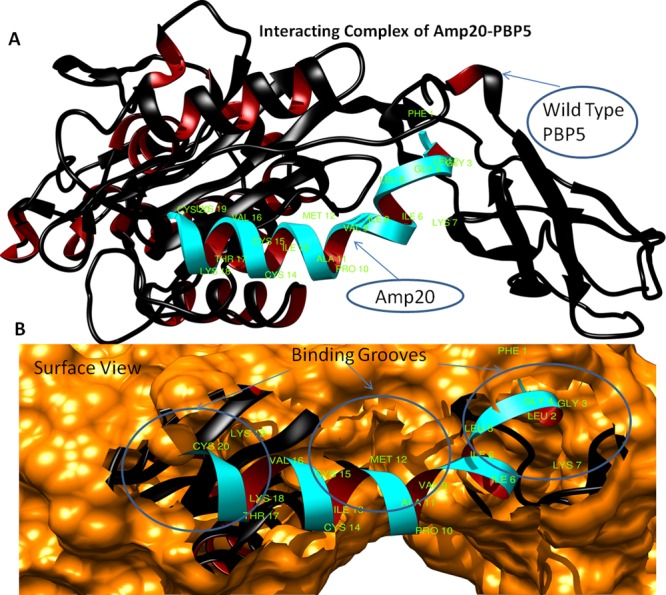
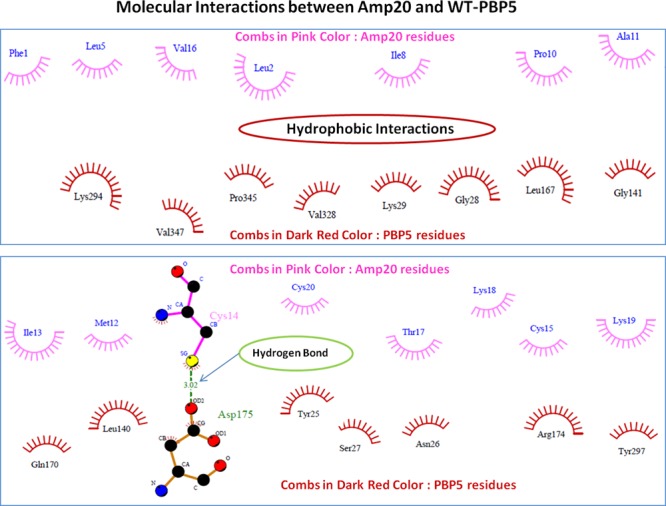
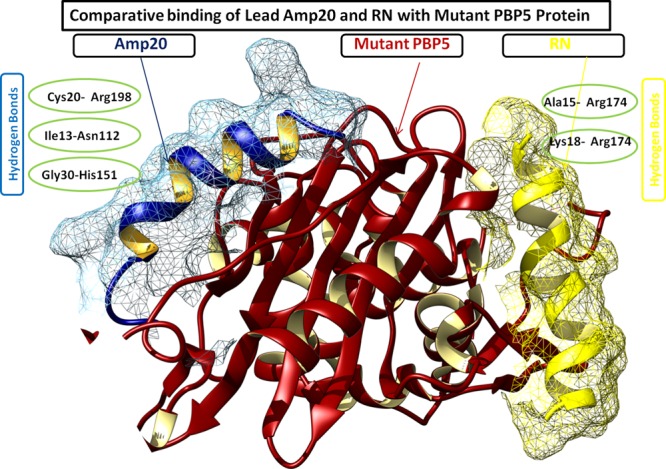
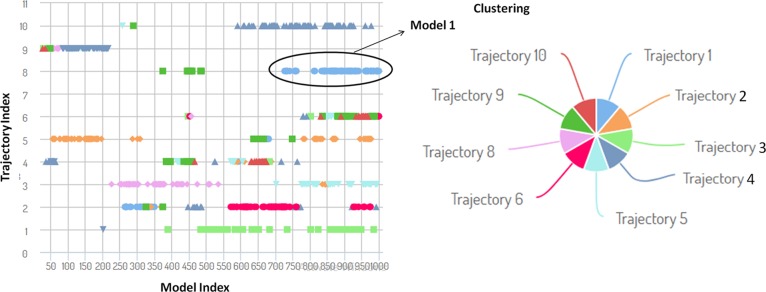
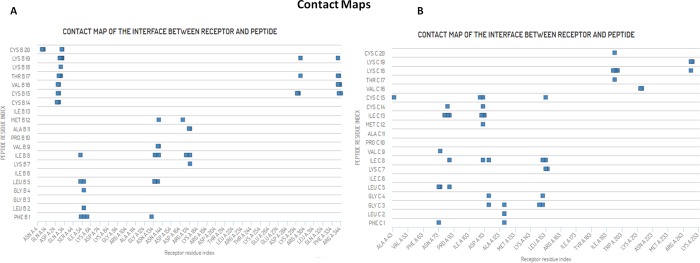
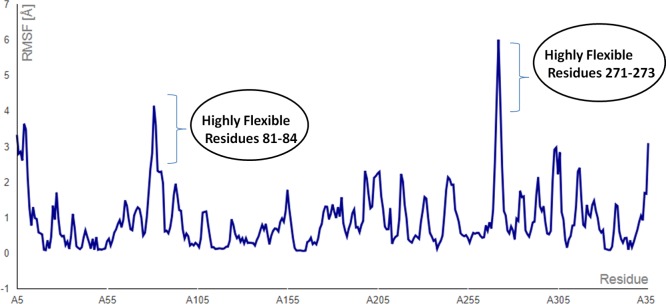
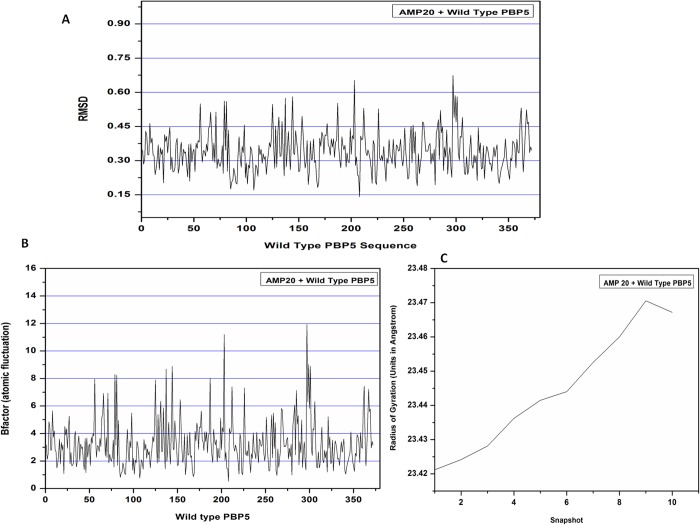
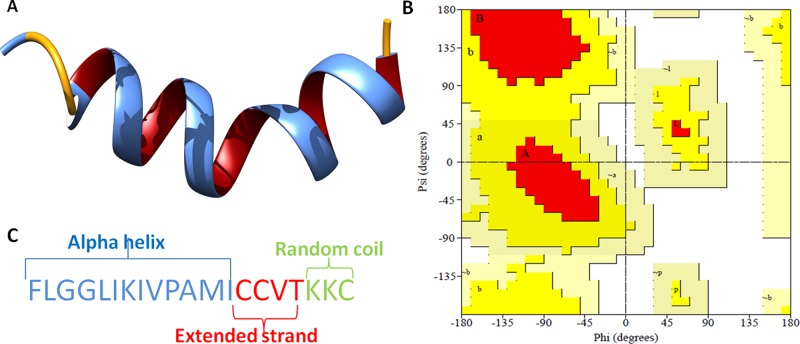
The mankind relies on the use of antibiotics for a healthy life. The epidemic-like emergence of drug-resistant bacterial strains is increasingly becoming one of the leading causes of morbidity and mortality, which gives rise to design a potential antimicrobial peptide (AMP). Here, we have designed the potential AMP using the extensive dynamics simulation since protein-peptide interactions are linked to large conformational changes. Therefore, we have employed the advanced computational avenue CABS molecular docking method that enabled the flexible peptide-protein molecular docking with a large-scale rearrangement of the protein. Lead AMP was investigated against the wild-type (WT) and mutant-PBP5 (MT-PBP5) proteins (antiresistance property). AMP20 showed strong interactions with wtPBP5 and mtPBP5 and involvement of a large number of elements in interactions determined through an atomic model study. Full flexibility analysis showed the stable interaction of AMP20 with both the wild-type and mutant form of PBP5 with root-mean-square deviation (RMSD) values of ∼4.51 and 4.85 Å, respectively. Moreover, peptide dynamics showed involvement of all residues of AMP20 through contact map analysis, and extensive simulation confirmed the stable interaction of AMP20, with lower values of RMSD, radius of gyration, and root-mean-square fluctuation. This study paves the way for a potential approach to design the AMP with amino acid walking and large-scale conformational rearrangements of amino acids.
Copyright © 2019 American Chemical Society.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
References
-
- O’Neill J. I. M. Antimicrobial Resistance: Tackling a crisis for the health and wealth of nations. Rev. Antimicrob. Resist. 2014, 1–16.
LinkOut - more resources
Full Text Sources