

tmap: an integrative framework based on topological data analysis for population-scale microbiome stratification and association studies
- PMID: 31870407
- PMCID: PMC6927166
- DOI: 10.1186/s13059-019-1871-4
tmap: an integrative framework based on topological data analysis for population-scale microbiome stratification and association studies
Abstract
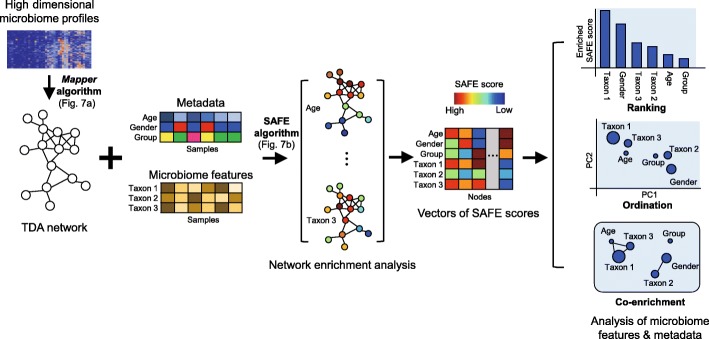
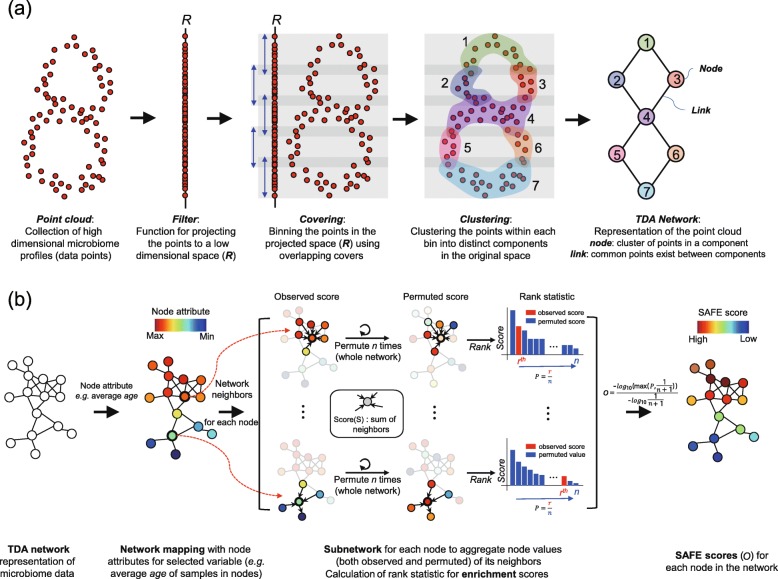
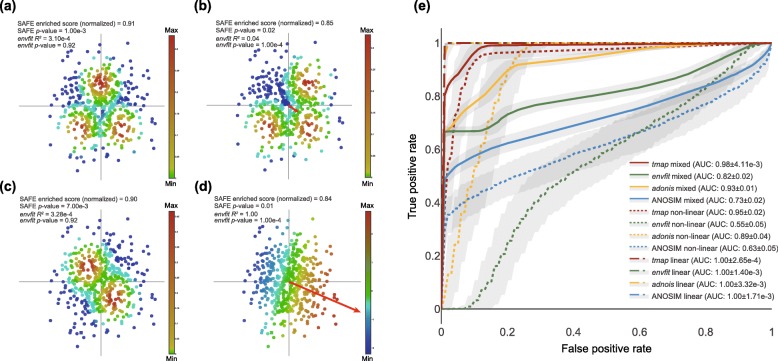
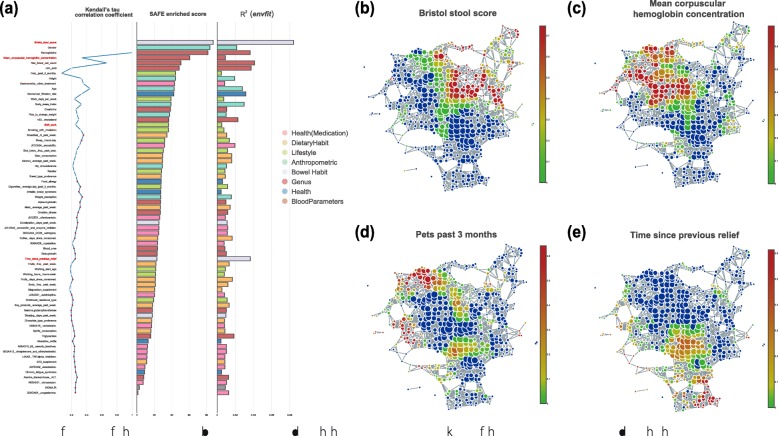
Untangling the complex variations of microbiome associated with large-scale host phenotypes or environment types challenges the currently available analytic methods. Here, we present tmap, an integrative framework based on topological data analysis for population-scale microbiome stratification and association studies. The performance of tmap in detecting nonlinear patterns is validated by different scenarios of simulation, which clearly demonstrate its superiority over the most commonly used methods. Application of tmap to several population-scale microbiomes extensively demonstrates its strength in revealing microbiome-associated host or environmental features and in understanding the systematic interrelations among their association patterns. tmap is available at https://github.com/GPZ-Bioinfo/tmap.
Keywords: Enterotype analysis; Microbiome stratification; Microbiome-wide association analysis; Nonlinear association; Population-scale microbiome; Topological data analysis.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
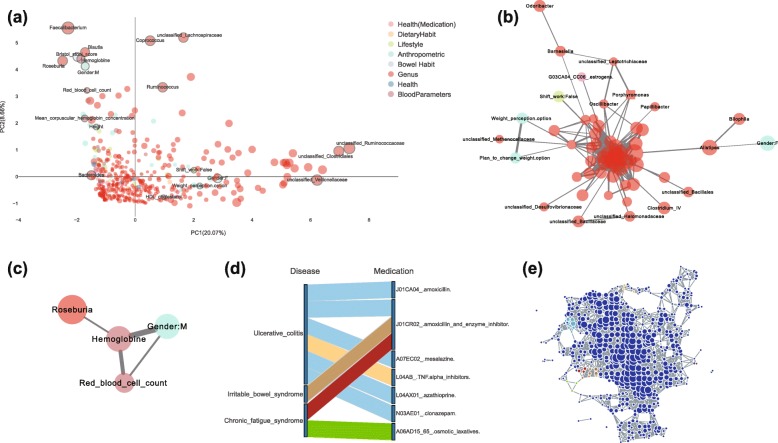
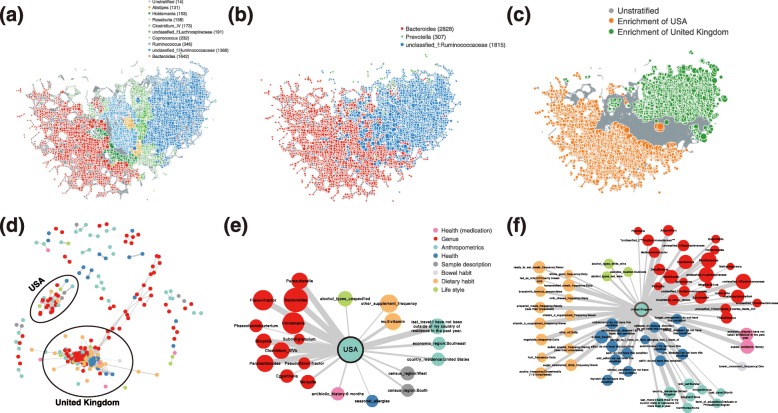
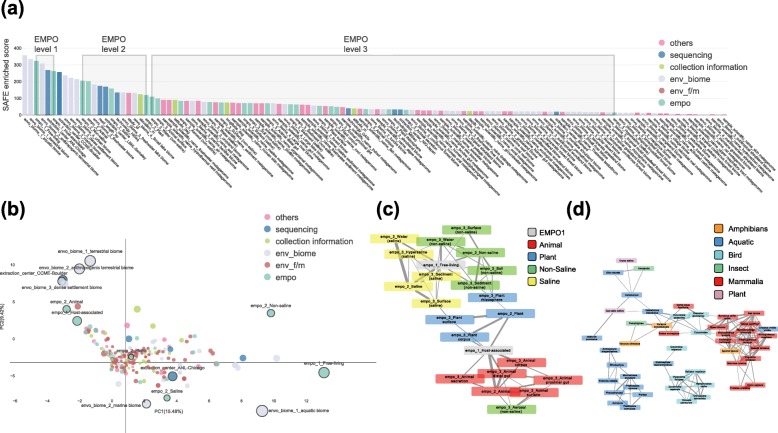
References
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
