

HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING
- PMID: 31874109
- PMCID: PMC7108911
- DOI: 10.1172/JCI129497
HPV16 drives cancer immune escape via NLRX1-mediated degradation of STING
Abstract
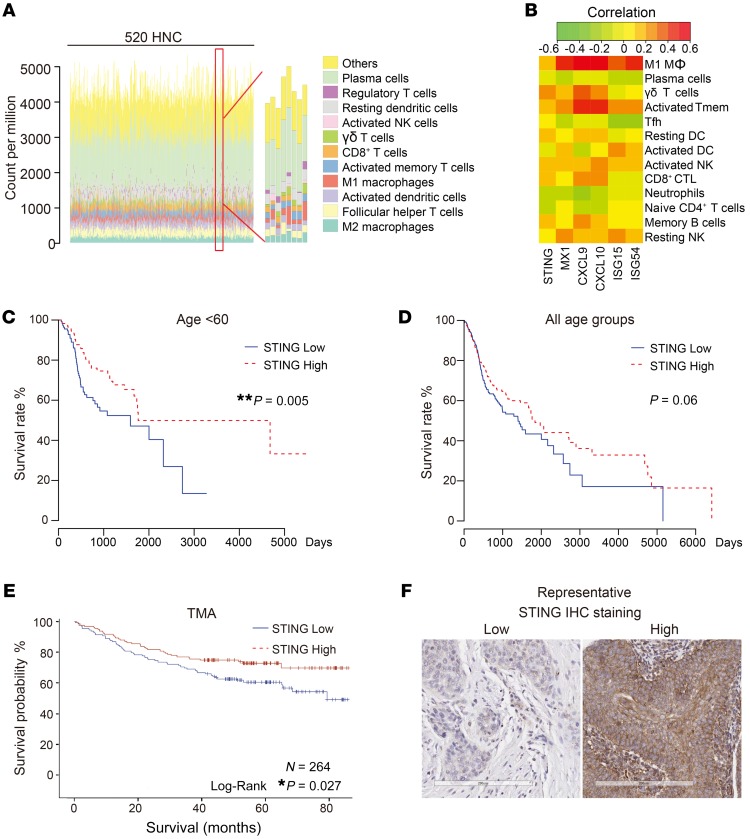
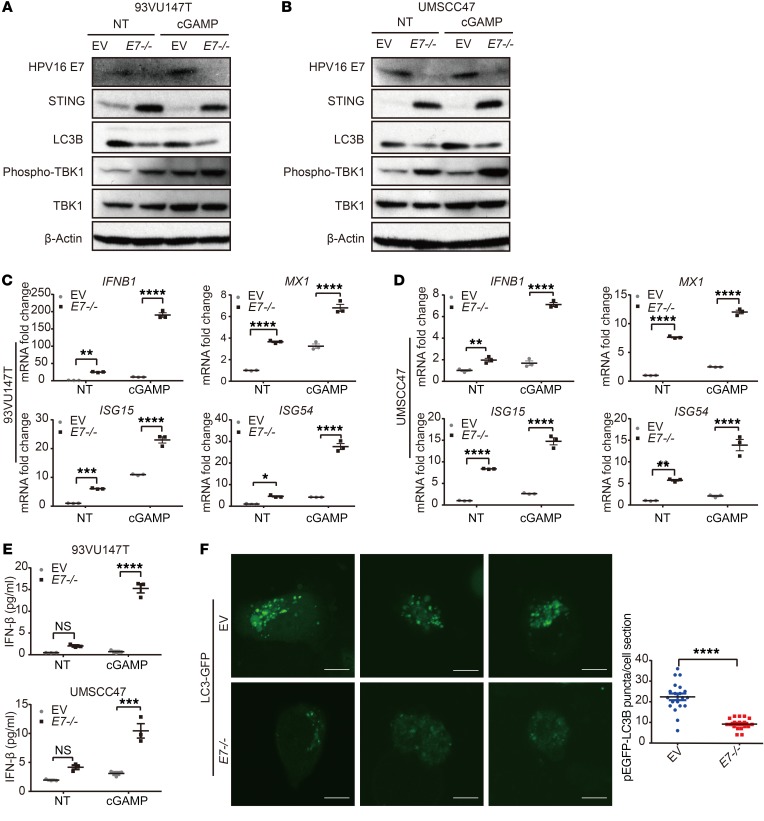
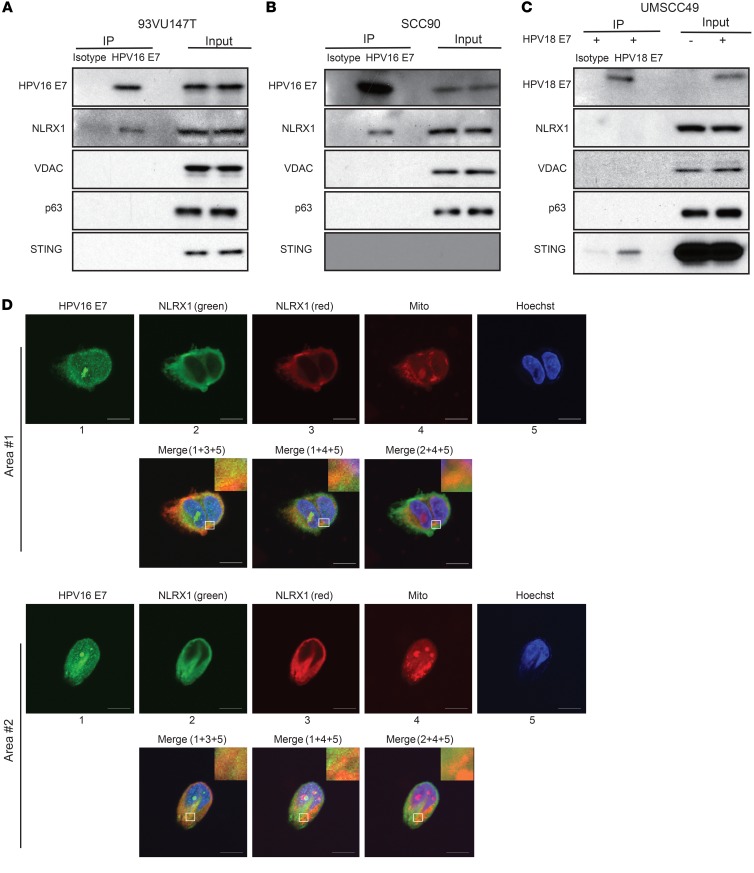
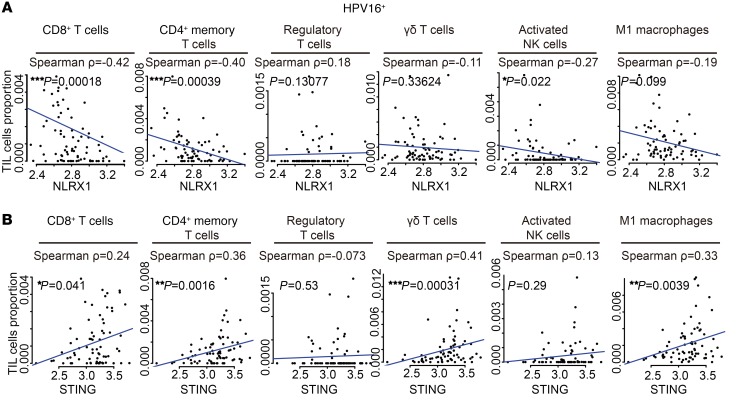
The incidence of human papillomavirus-positive (HPV+) head and neck squamous cell carcinoma (HNSCC) has surpassed that of cervical cancer and is projected to increase rapidly until 2060. The coevolution of HPV with transforming epithelial cells leads to the shutdown of host immune detection. Targeting proximal viral nucleic acid-sensing machinery is an evolutionarily conserved strategy among viruses to enable immune evasion. However, E7 from the dominant HPV subtype 16 in HNSCC shares low homology with HPV18 E7, which was shown to inhibit the STING DNA-sensing pathway. The mechanisms by which HPV16 suppresses STING remain unknown. Recently, we characterized the role of the STING/type I interferon (IFN-I) pathway in maintaining immunogenicity of HNSCC in mouse models. Here we extended those findings into the clinical domain using tissue microarrays and machine learning-enhanced profiling of STING signatures with immune subsets. We additionally showed that HPV16 E7 uses mechanisms distinct from those used by HPV18 E7 to antagonize the STING pathway. We identified NLRX1 as a critical intermediary partner to facilitate HPV16 E7-potentiated STING turnover. The depletion of NLRX1 resulted in significantly improved IFN-I-dependent T cell infiltration profiles and tumor control. Overall, we discovered a unique HPV16 viral strategy to thwart host innate immune detection that can be further exploited to restore cancer immunogenicity.
Keywords: Autophagy; Head and neck cancer; Immunology; Innate immunity; Oncology.
Conflict of interest statement
Figures






References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
