

The somatic mutation landscape of the human body
- PMID: 31874648
- PMCID: PMC6930685
- DOI: 10.1186/s13059-019-1919-5
The somatic mutation landscape of the human body
Abstract
Background: Somatic mutations in healthy tissues contribute to aging, neurodegeneration, and cancer initiation, yet they remain largely uncharacterized.
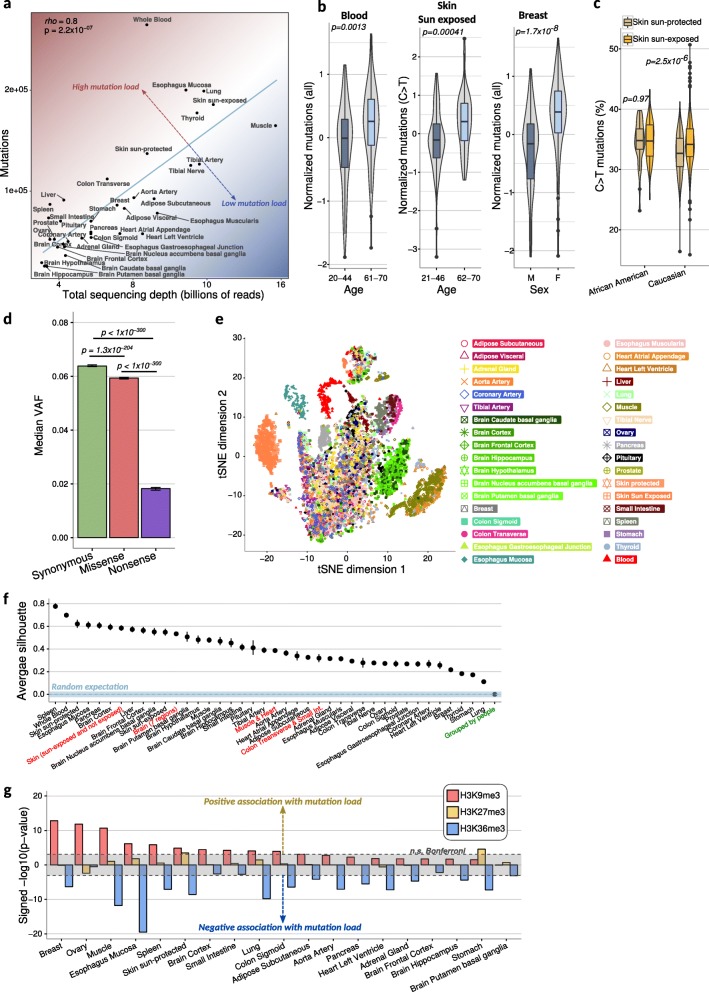
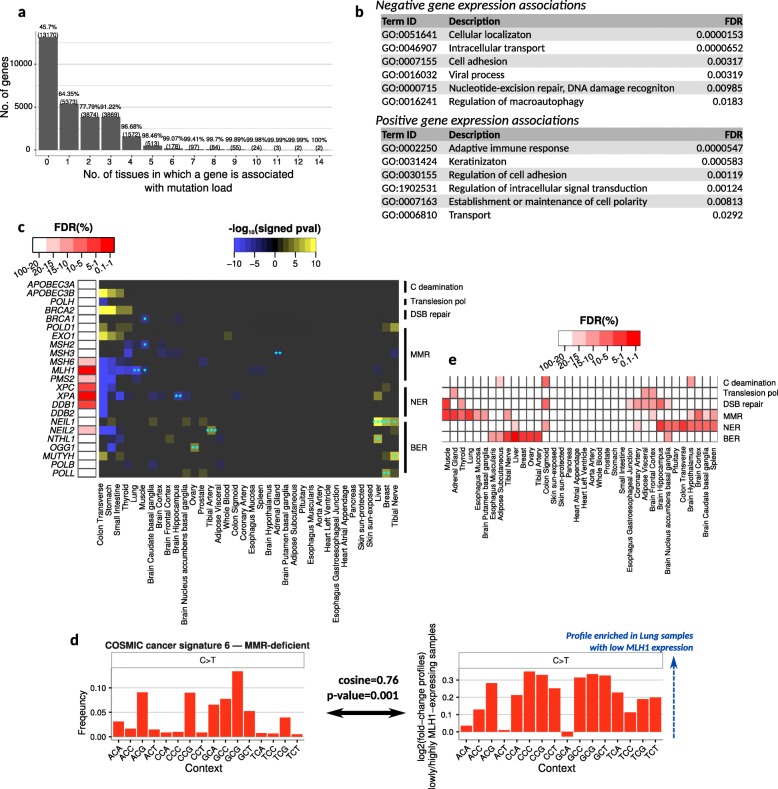
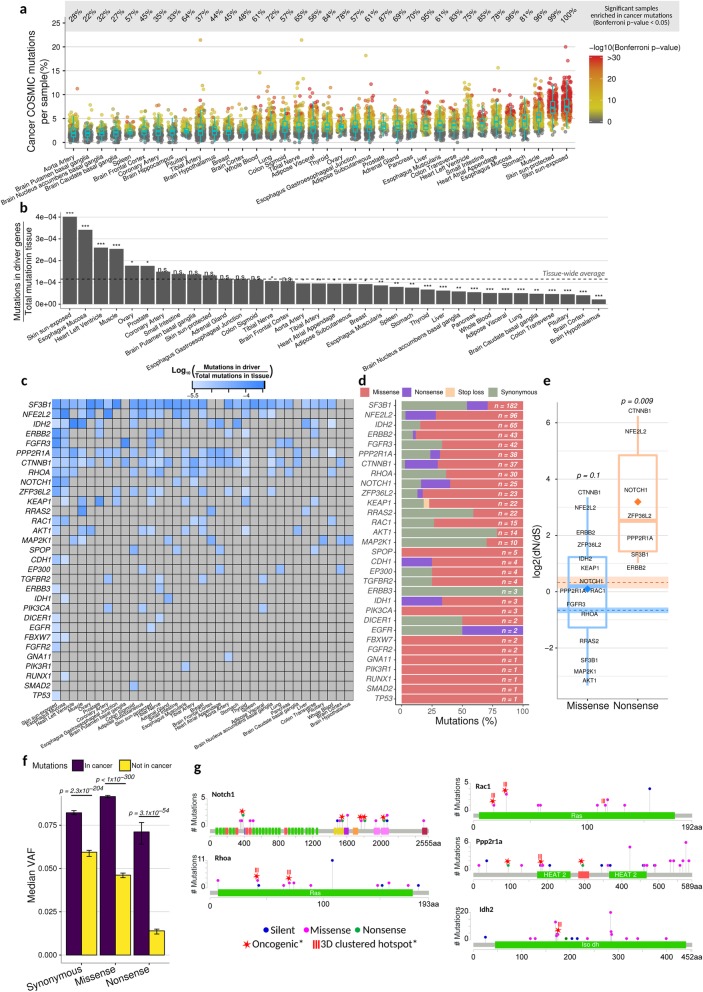
Results: To gain a better understanding of the genome-wide distribution and functional impact of somatic mutations, we leverage the genomic information contained in the transcriptome to uniformly call somatic mutations from over 7500 tissue samples, representing 36 distinct tissues. This catalog, containing over 280,000 mutations, reveals a wide diversity of tissue-specific mutation profiles associated with gene expression levels and chromatin states. For example, lung samples with low expression of the mismatch-repair gene MLH1 show a mutation signature of deficient mismatch repair. In addition, we find pervasive negative selection acting on missense and nonsense mutations, except for mutations previously observed in cancer samples, which are under positive selection and are highly enriched in many healthy tissues.
Conclusions: These findings reveal fundamental patterns of tissue-specific somatic evolution and shed light on aging and the earliest stages of tumorigenesis.
Keywords: Aging; Cancer; Genomic instability; Human; Somatic evolution; Somatic mutation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
