

Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease
- PMID: 31874800
- PMCID: PMC7218796
- DOI: 10.1016/j.kint.2019.08.038
Detection and characterization of mosaicism in autosomal dominant polycystic kidney disease
Abstract
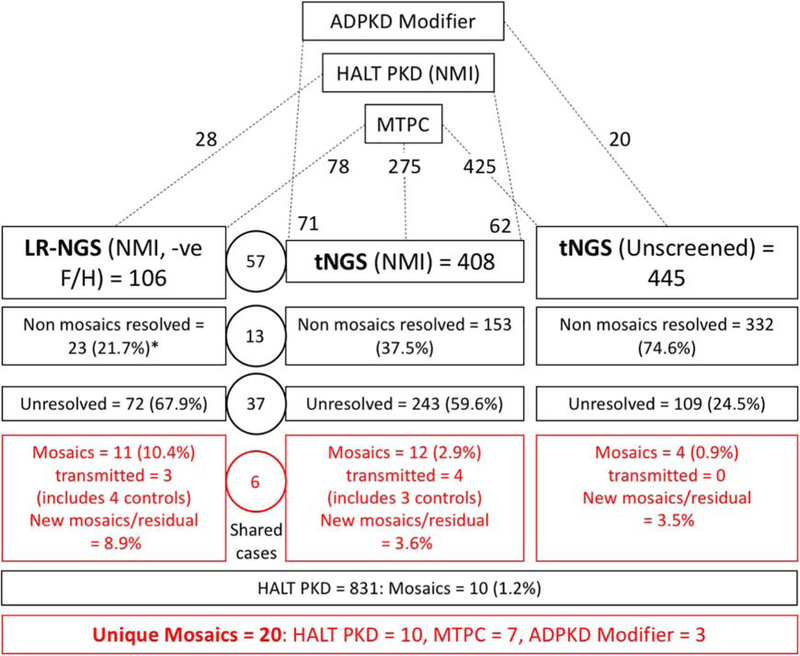
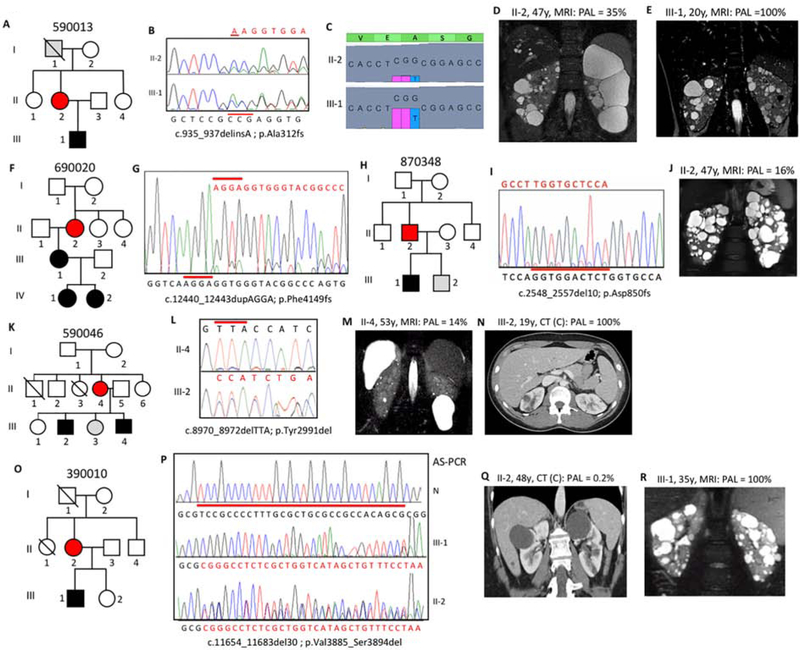
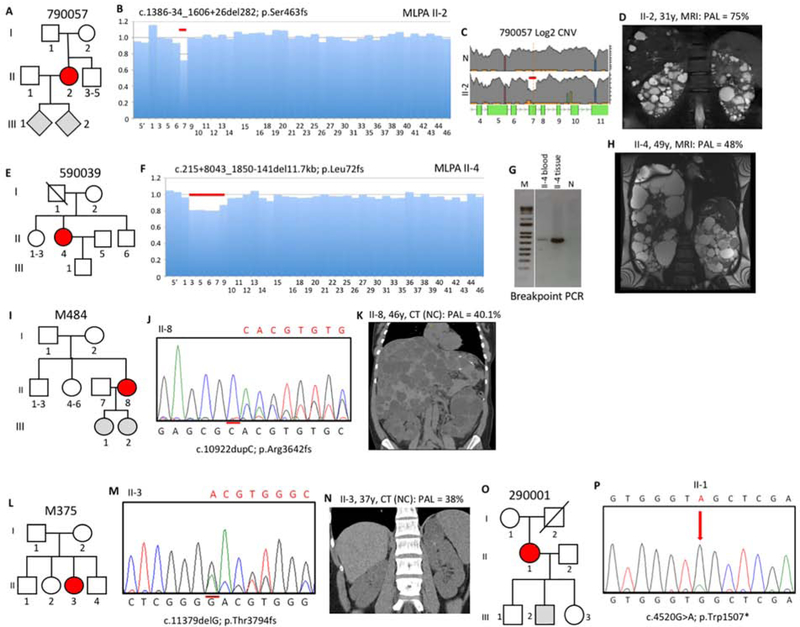
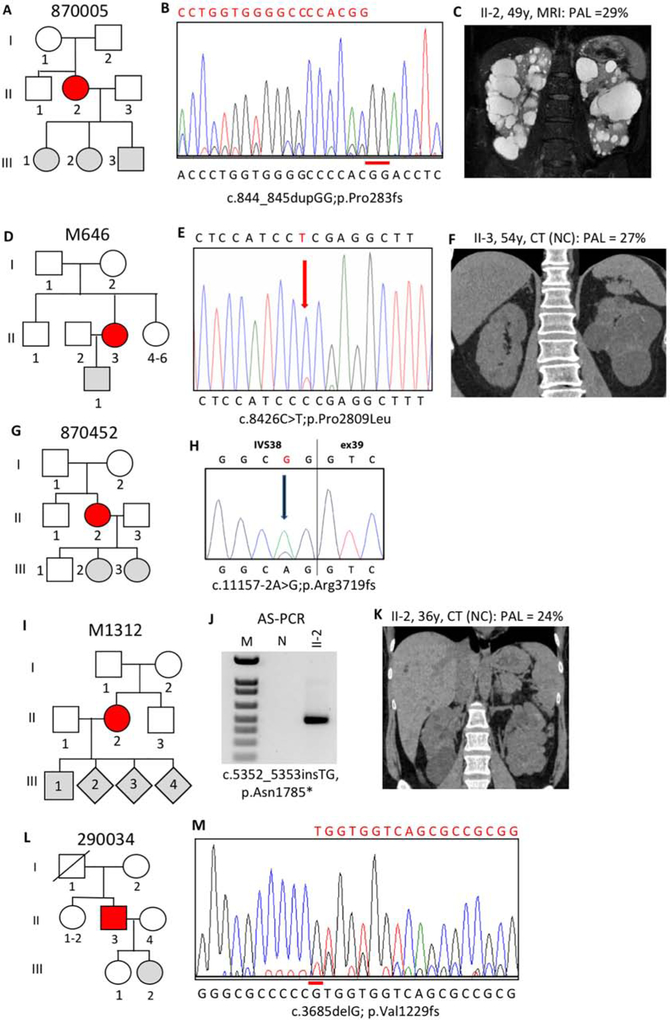
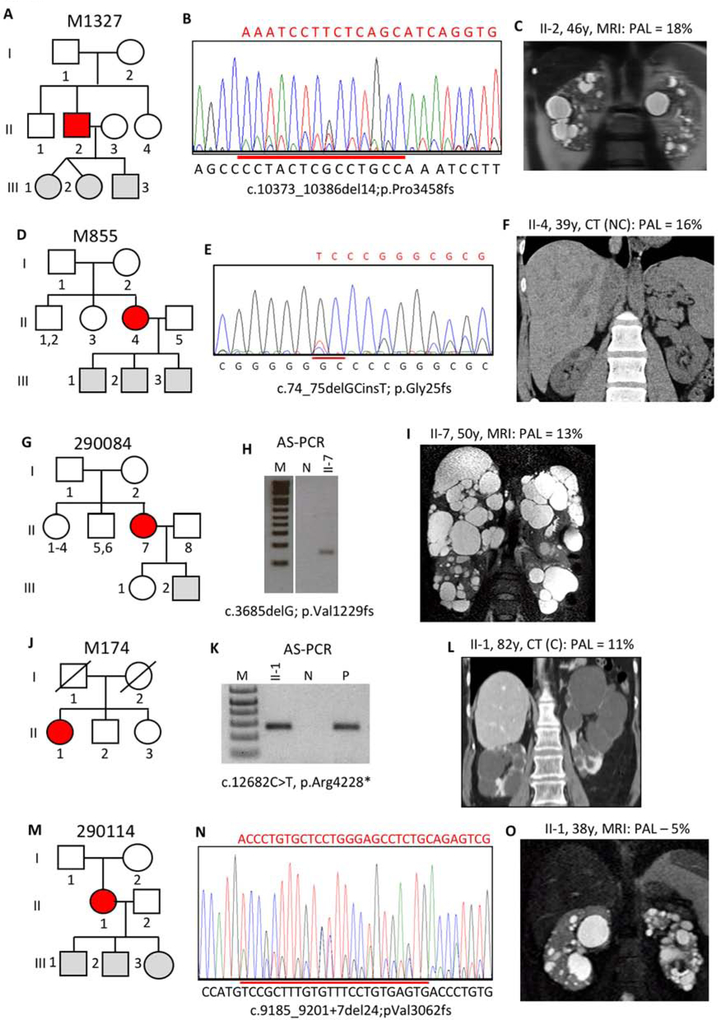
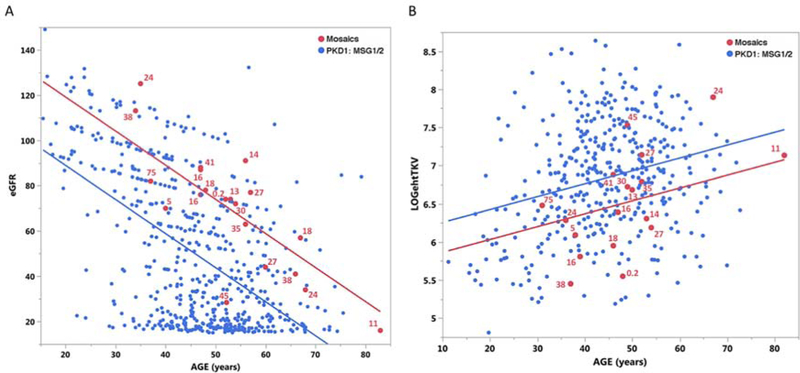
Autosomal dominant polycystic kidney disease (ADPKD) is an inherited, progressive nephropathy accounting for 4-10% of end stage renal disease worldwide. PKD1 and PKD2 are the most common disease loci, but even accounting for other genetic causes, about 7% of families remain unresolved. Typically, these unsolved cases have relatively mild kidney disease and often have a negative family history. Mosaicism, due to de novo mutation in the early embryo, has rarely been identified by conventional genetic analysis of ADPKD families. Here we screened for mosaicism by employing two next generation sequencing screens, specific analysis of PKD1 and PKD2 employing long-range polymerase chain reaction, or targeted capture of cystogenes. We characterized mosaicism in 20 ADPKD families; the pathogenic variant was transmitted to the next generation in five families and sporadic in 15. The mosaic pathogenic variant was newly discovered by next generation sequencing in 13 families, and these methods precisely quantified the level of mosaicism in all. All of the mosaic cases had PKD1 mutations, 14 were deletions or insertions, and 16 occurred in females. Analysis of kidney size and function showed the mosaic cases had milder disease than a control PKD1 population, but only a few had clearly asymmetric disease. Thus, in a typical ADPKD population, readily detectable mosaicism by next generation sequencing accounts for about 1% of cases, and about 10% of genetically unresolved cases with an uncertain family history. Hence, identification of mosaicism is important to fully characterize ADPKD populations and provides informed prognostic information.
Keywords: ADPKD; PKD1; diagnostics; genotype/phenotype correlations; mosaicism; mutations; prognostics.
Copyright © 2019 International Society of Nephrology. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Disclosure
All the authors declared no competing interests.
Figures
Comment in
-
Next-generation sequencing for detection of somatic mosaicism in autosomal dominant polycystic kidney disease.Kidney Int. 2020 Feb;97(2):261-263. doi: 10.1016/j.kint.2019.11.019. Kidney Int. 2020. PMID: 31980075
References
-
- Torres VE, Harris PC, Pirson Y. Autosomal dominant polycystic kidney disease. Lancet 2007; 369: 1287–1301. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- U01 DK062401/DK/NIDDK NIH HHS/United States
- M01 RR000585/RR/NCRR NIH HHS/United States
- P30 DK090868/DK/NIDDK NIH HHS/United States
- U01 DK062402/DK/NIDDK NIH HHS/United States
- UL1 RR025780/RR/NCRR NIH HHS/United States
- UL1 TR001064/TR/NCATS NIH HHS/United States
- UL1 RR025752/RR/NCRR NIH HHS/United States
- UL1 TR000001/TR/NCATS NIH HHS/United States
- M01 RR001032/RR/NCRR NIH HHS/United States
- UL1 TR000135/TR/NCATS NIH HHS/United States
- U01 DK062408/DK/NIDDK NIH HHS/United States
- UL1 TR000454/TR/NCATS NIH HHS/United States
- M01 RR000051/RR/NCRR NIH HHS/United States
- U01 DK062411/DK/NIDDK NIH HHS/United States
- UL1 RR025008/RR/NCRR NIH HHS/United States
- U01 DK062410/DK/NIDDK NIH HHS/United States
- R01 DK058816/DK/NIDDK NIH HHS/United States
- P30 DK090728/DK/NIDDK NIH HHS/United States
- R01 DK079856/DK/NIDDK NIH HHS/United States
- M01 RR000039/RR/NCRR NIH HHS/United States
- UL1 RR033179/RR/NCRR NIH HHS/United States
- M01 RR000054/RR/NCRR NIH HHS/United States
- M01 RR023940/RR/NCRR NIH HHS/United States
- R01 DK044863/DK/NIDDK NIH HHS/United States
- UL1 TR001082/TR/NCATS NIH HHS/United States
- UL1 TR001102/TR/NCATS NIH HHS/United States
- U01 DK082230/DK/NIDDK NIH HHS/United States
- UL1 RR024150/RR/NCRR NIH HHS/United States
- UL1 RR025758/RR/NCRR NIH HHS/United States
- U01 DK079856/DK/NIDDK NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
