

The genetics and genomics of cystic fibrosis
- PMID: 31879237
- PMCID: PMC8008819
- DOI: 10.1016/j.jcf.2019.11.003
The genetics and genomics of cystic fibrosis
Abstract
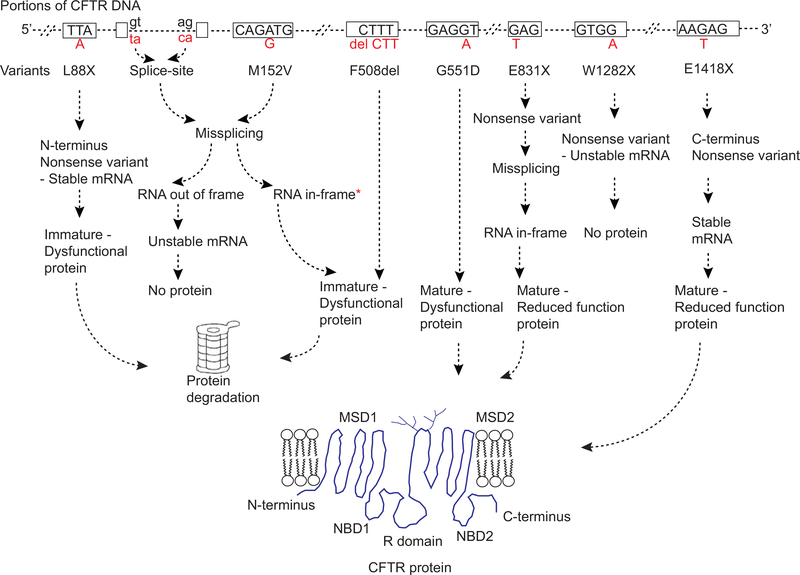
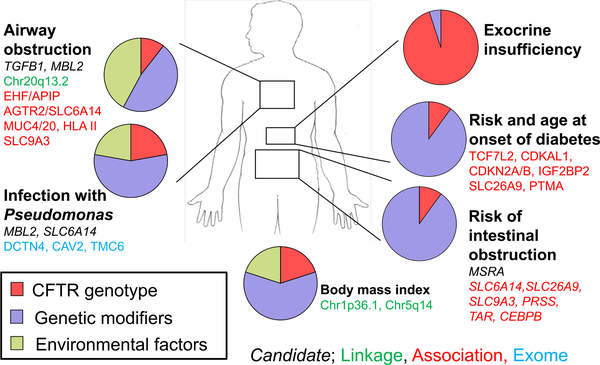
Genetics is the branch of biology concerned with study of individual genes and how they work whereas genomics is involved with the analysis of all genes and their interactions. Both of these approaches have been applied extensively to CF. Identification of the CFTR gene initiated the dissection of CF genetics at the molecular level. Subsequently, thousands of variants were found in the gene and the functional consequences of a subset have been studied in detail. The completion of the human genome ushered in a new phase of study where the role of genes beyond CFTR could be evaluated for their contribution to the severity of CF. This will be a brief overview of the contribution of these complementary methods to our understanding of CF pathogenesis.
Keywords: Association; CFTR; Cystic fibrosis; Genetic modifiers; Linkage.
Copyright © 2019 European Cystic Fibrosis Society. Published by Elsevier B.V. All rights reserved.
Conflict of interest statement
Declaration of Competing Interest None.
Figures
References
-
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, Chakravarti A, et al. Identification of the cystic fibrosis gene: genetic analysis. Science 1989;245:1073–80. - PubMed
-
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, Grzelczak Z, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science 1989;245(4922):1066–73. - PubMed
-
- Rommens JM, Iannuzzi MC, Kerem B, Drumm ML, Melmer G, Dean M, et al. Identification of the cystic fibrosis gene: chromosome walking and jumping. Science 1989;245:1059–65. - PubMed
-
- Amaral MD. Novel personalized therapies for cystic fibrosis: treating the basic defect in all patients. J Intern Med 2015;277(2):155–66. - PubMed
-
- De Boeck K, Amaral MD. Progress in therapies for cystic fibrosis. Lancet Respir Med 2016;4(8):662–74. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
