

Cutting into the Substrate Dominance: Pharmacophore and Structure-Based Approaches toward Inhibiting Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H
- PMID: 31880912
- PMCID: PMC7144833
- DOI: 10.1021/acs.accounts.9b00450
Cutting into the Substrate Dominance: Pharmacophore and Structure-Based Approaches toward Inhibiting Human Immunodeficiency Virus Reverse Transcriptase-Associated Ribonuclease H
Abstract
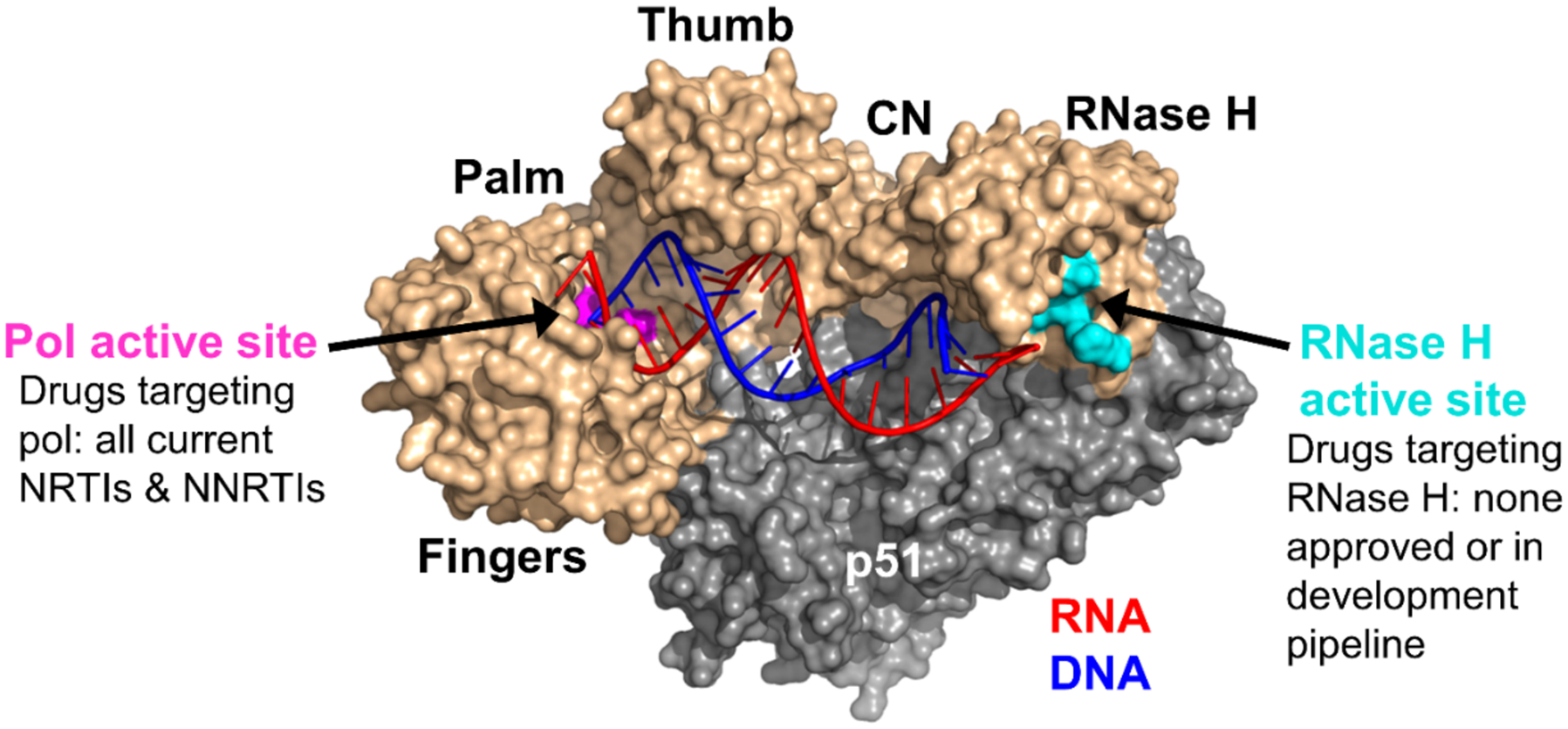
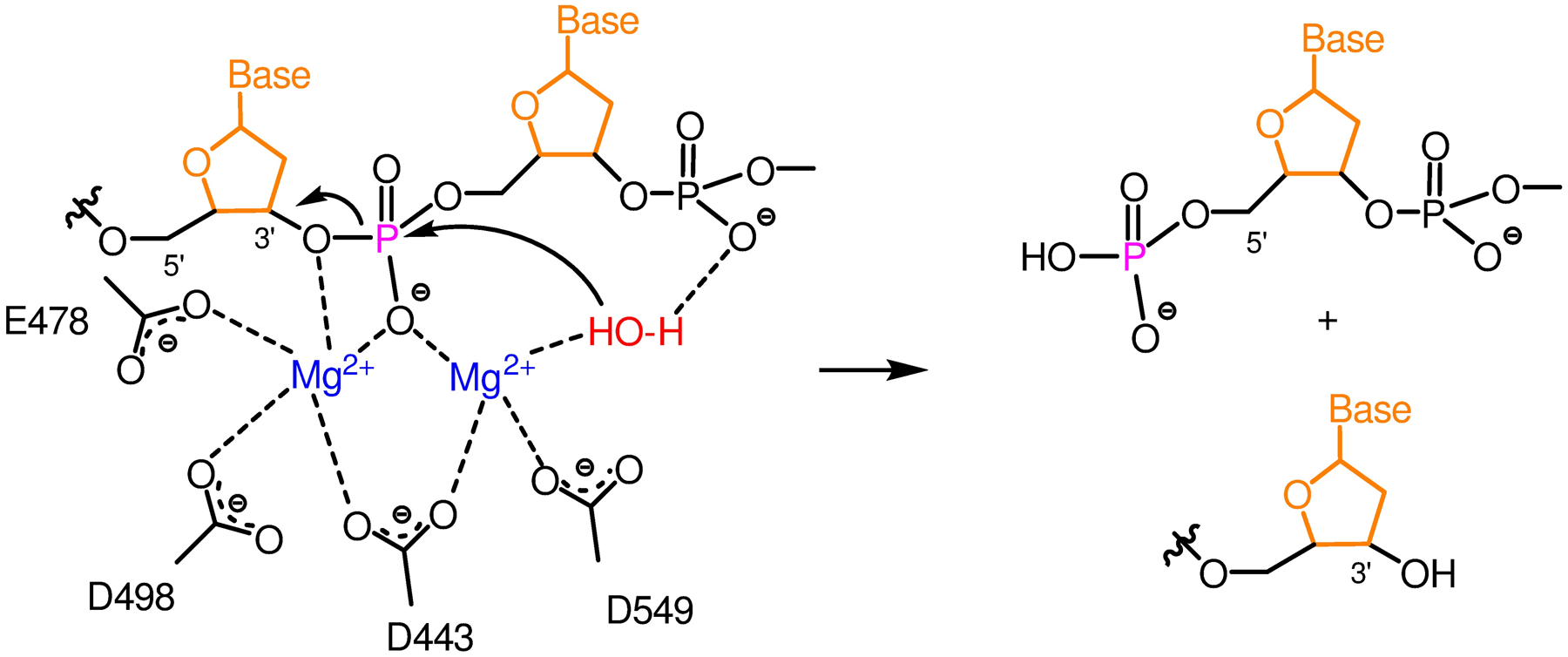
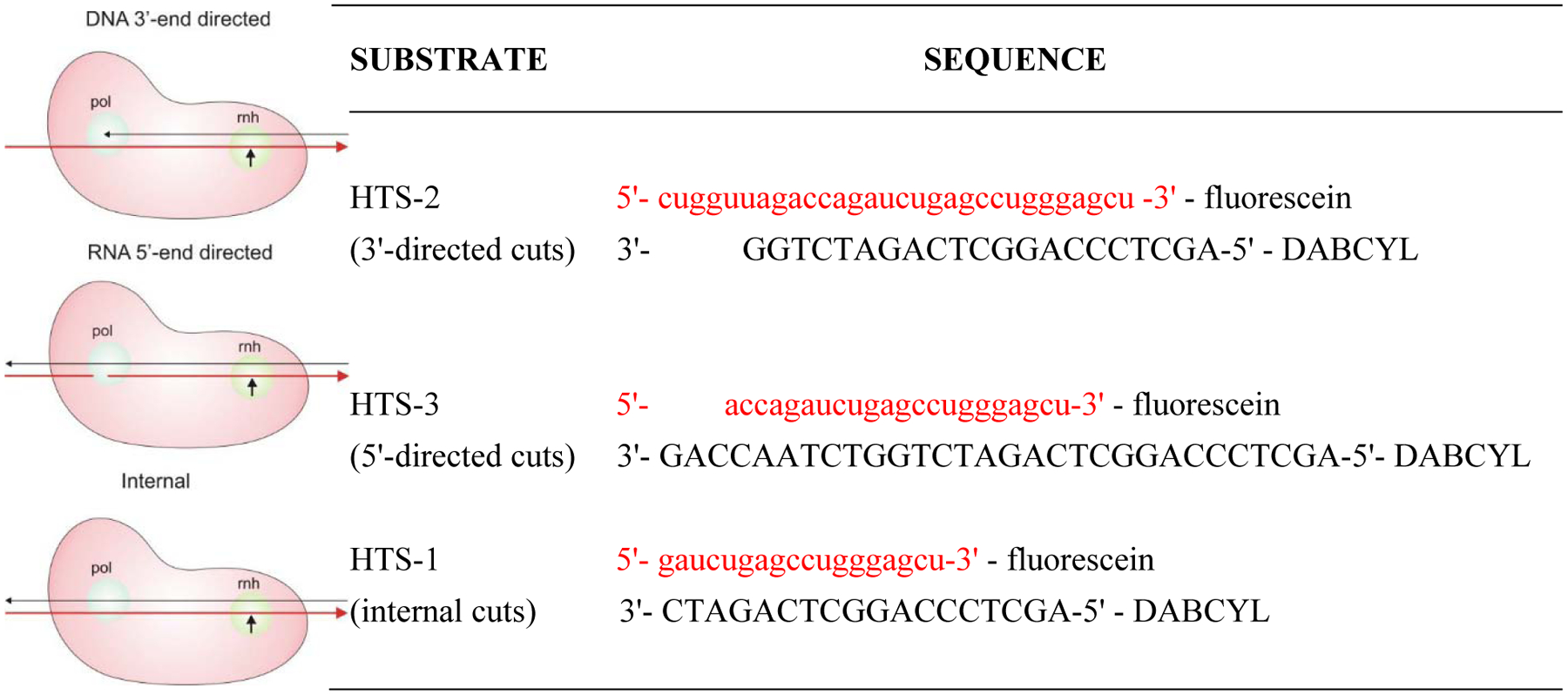
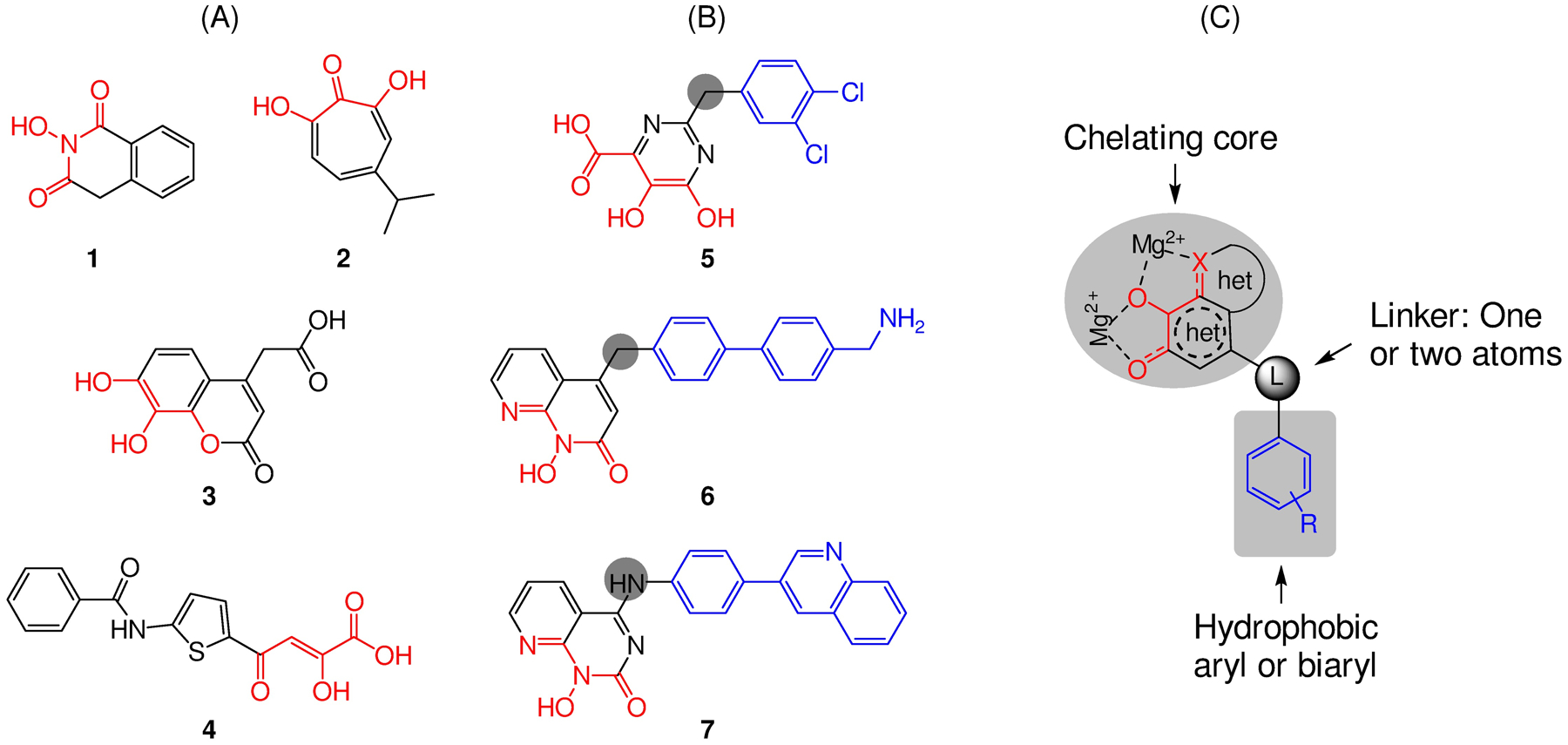
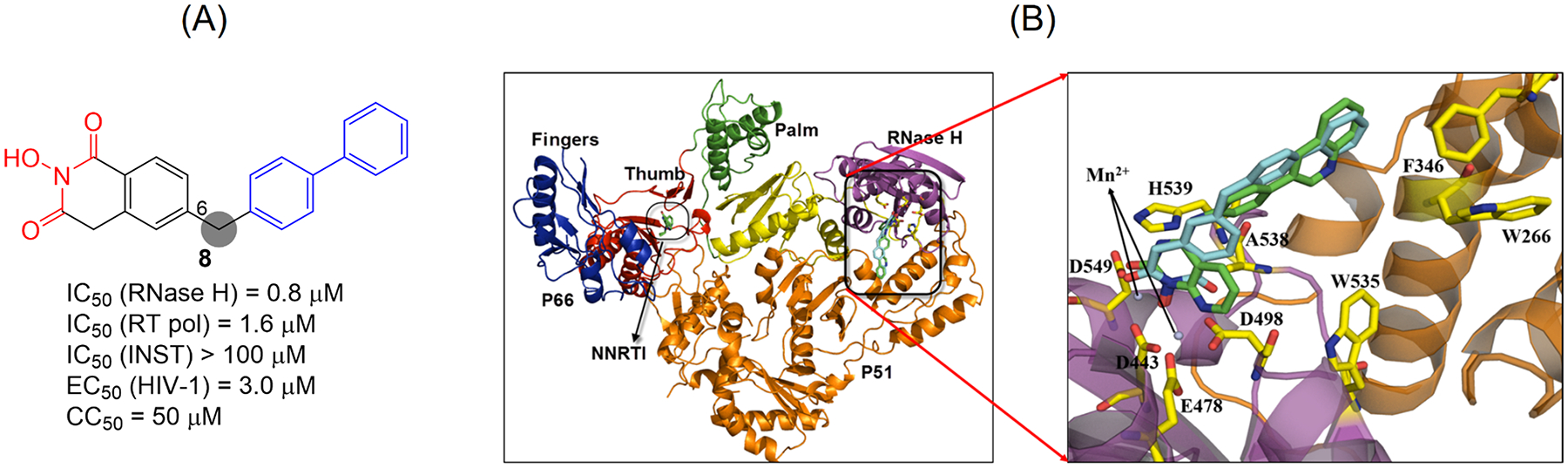
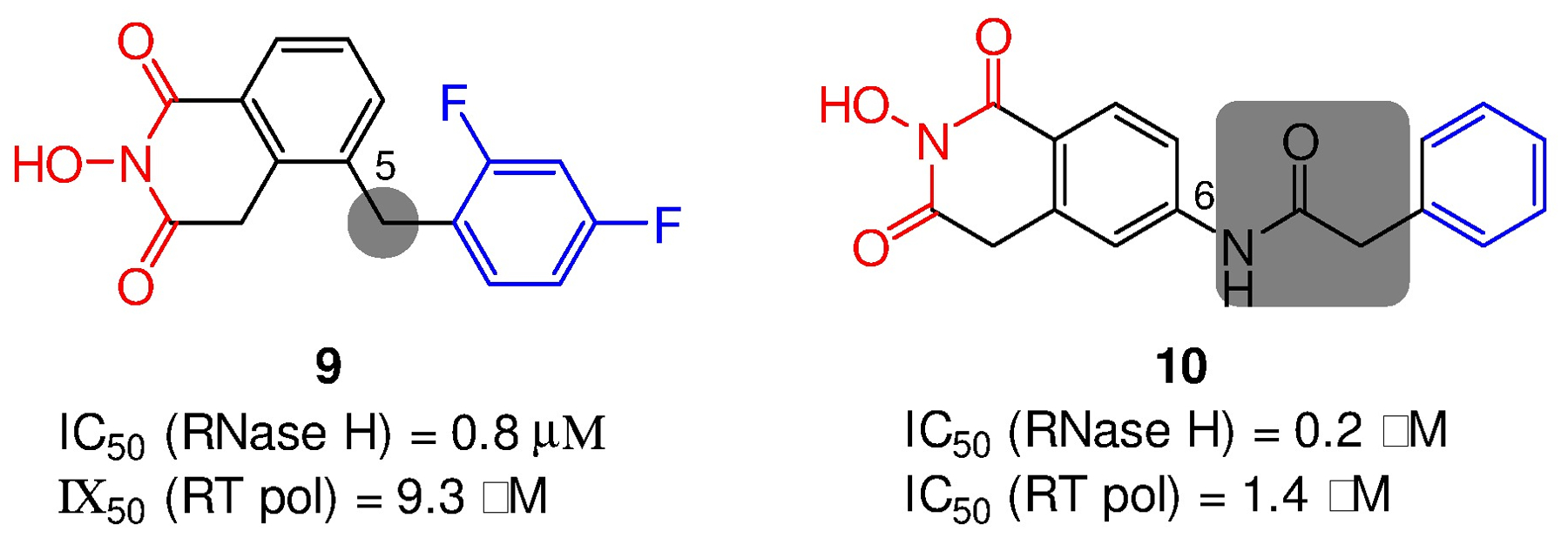
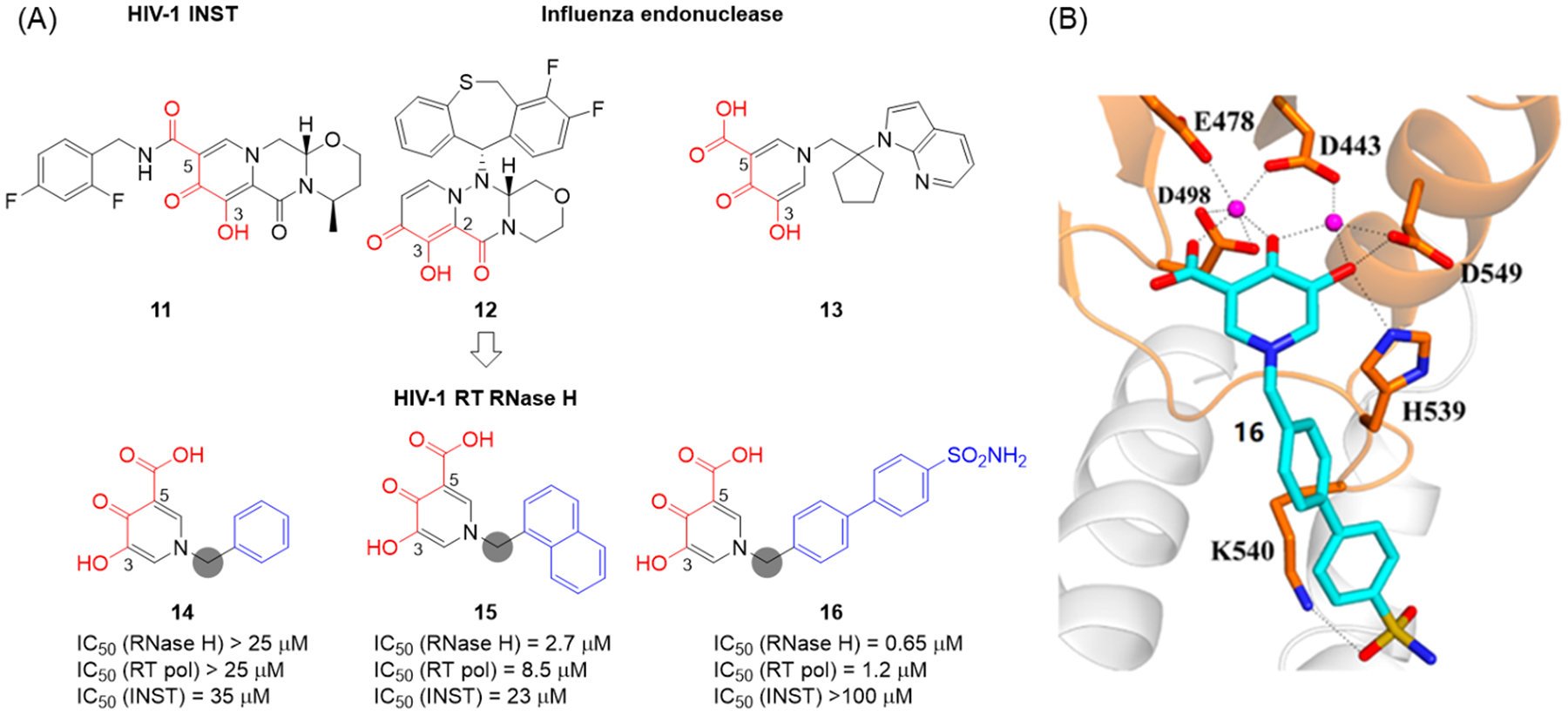
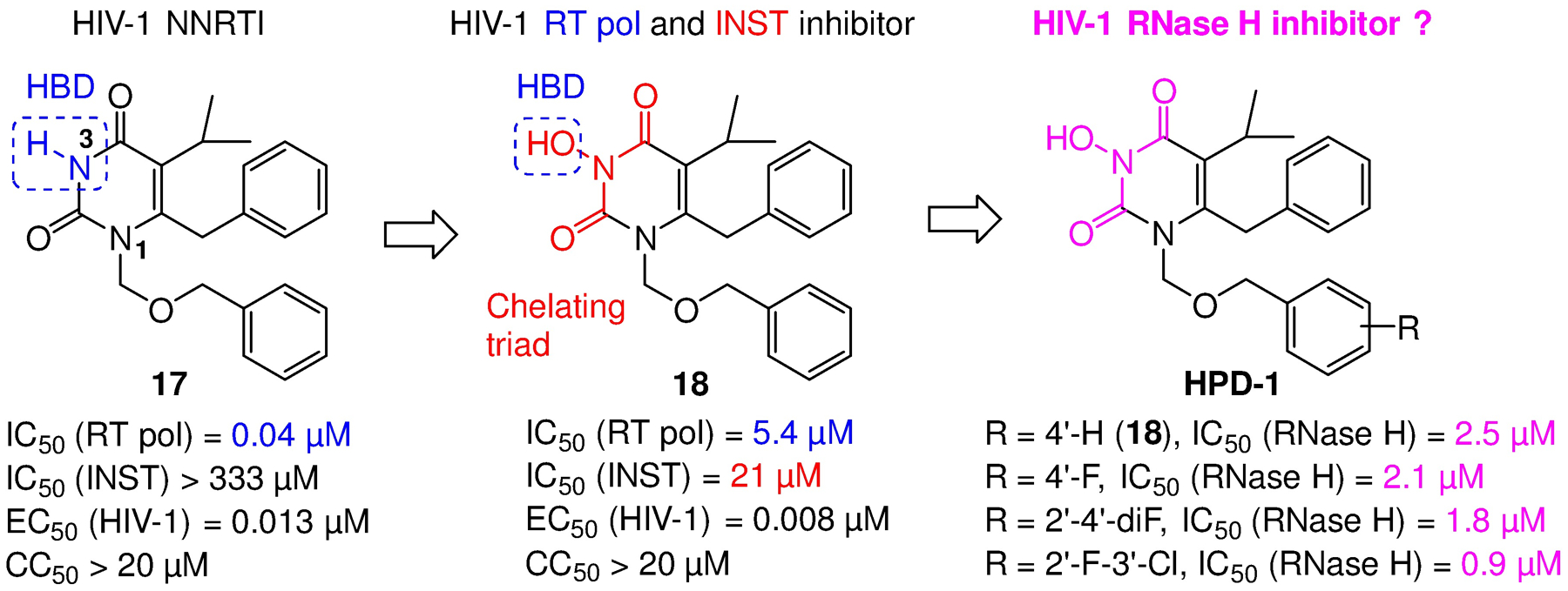
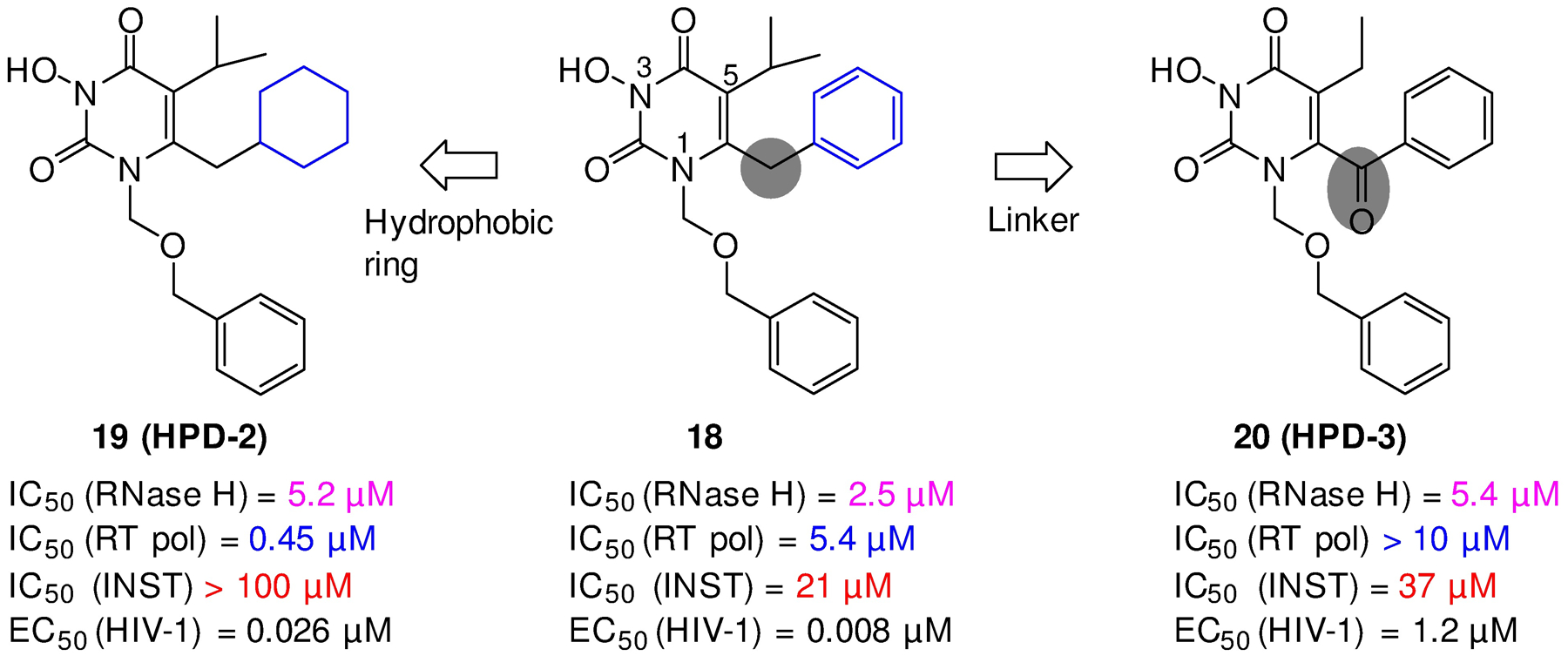
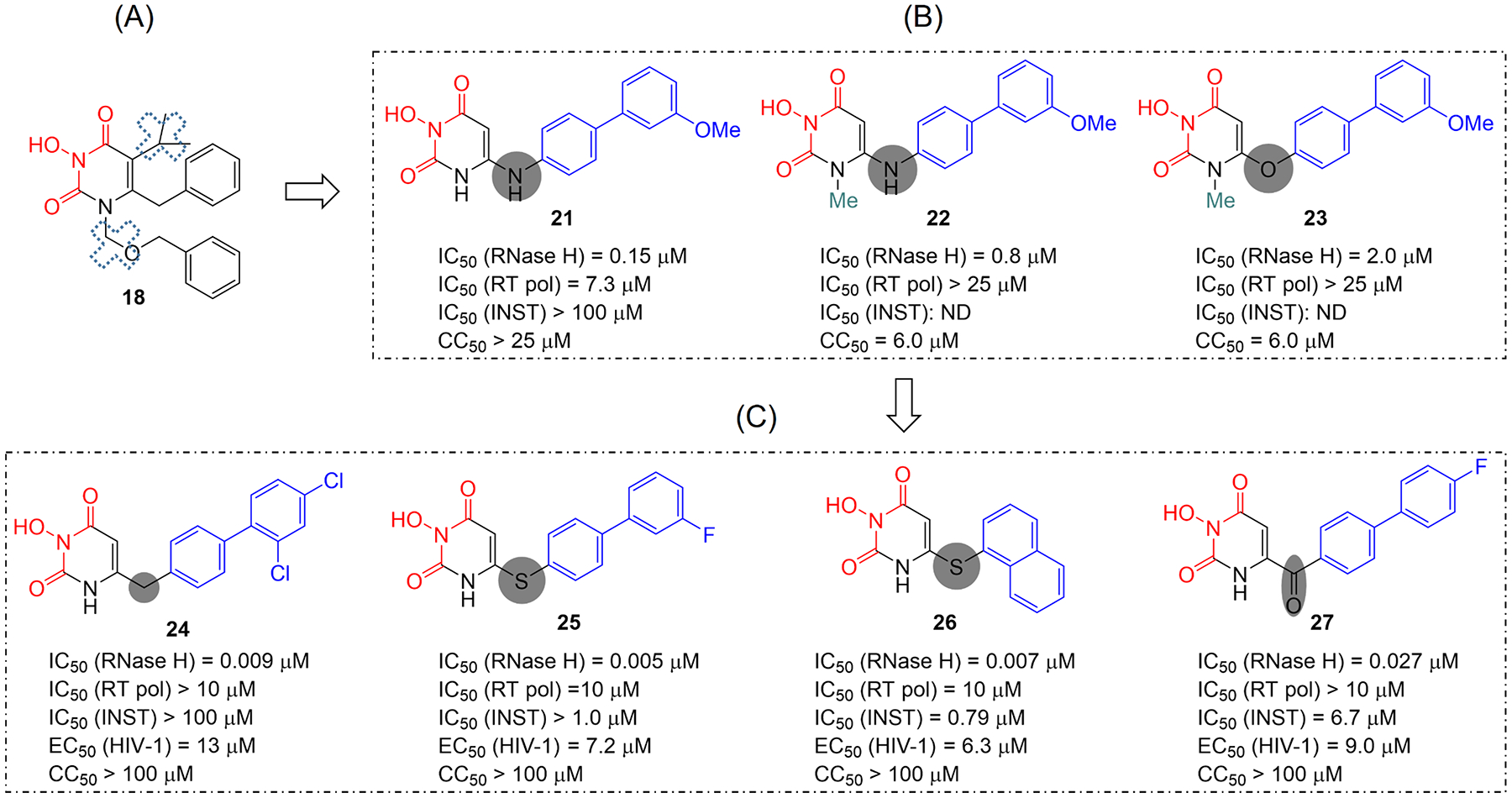
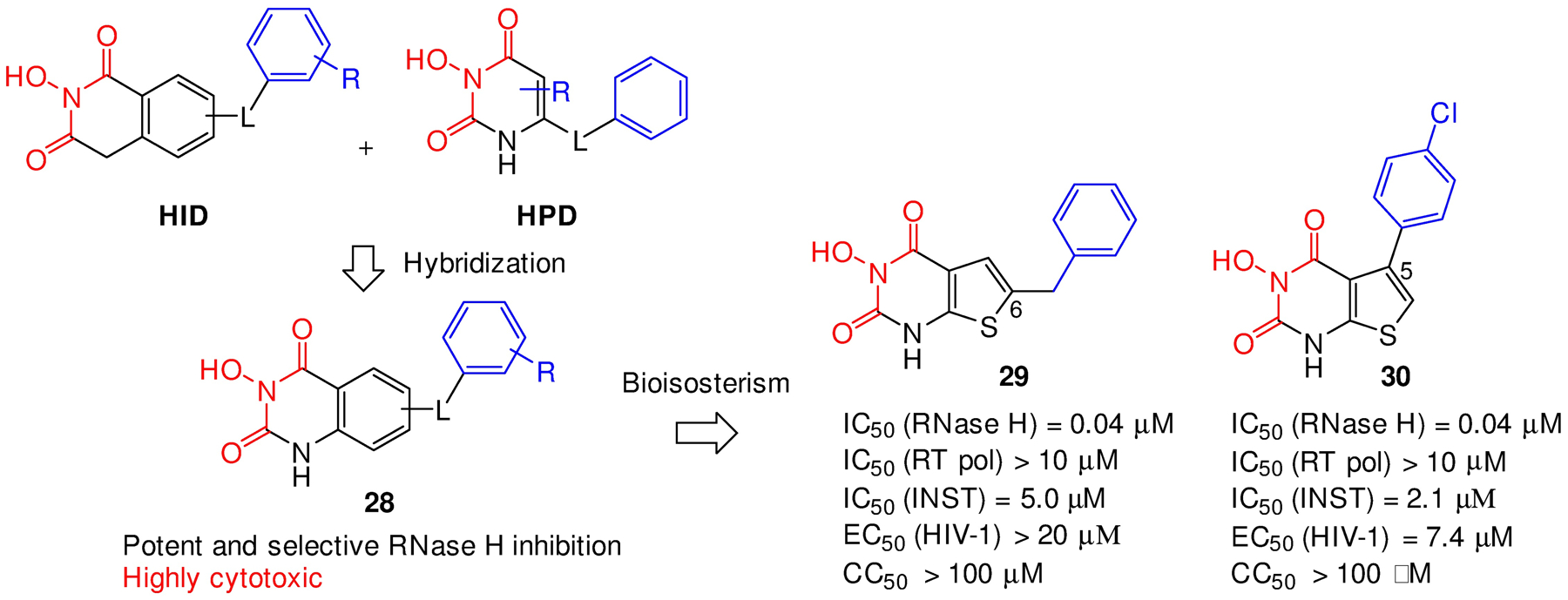
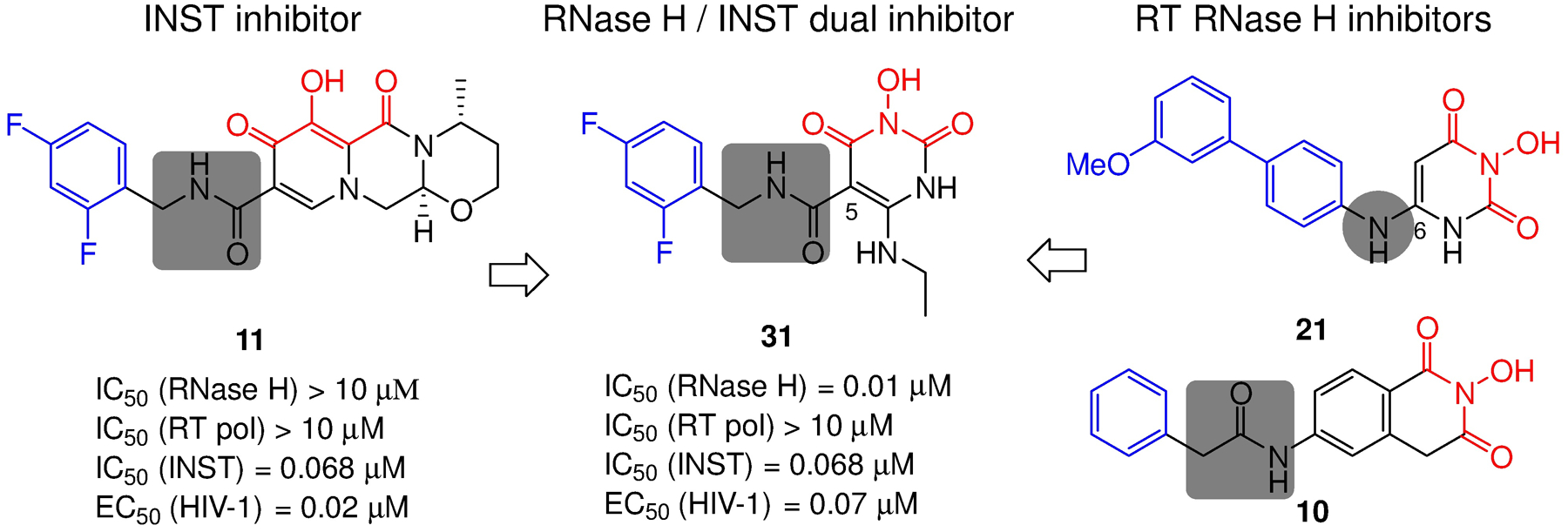
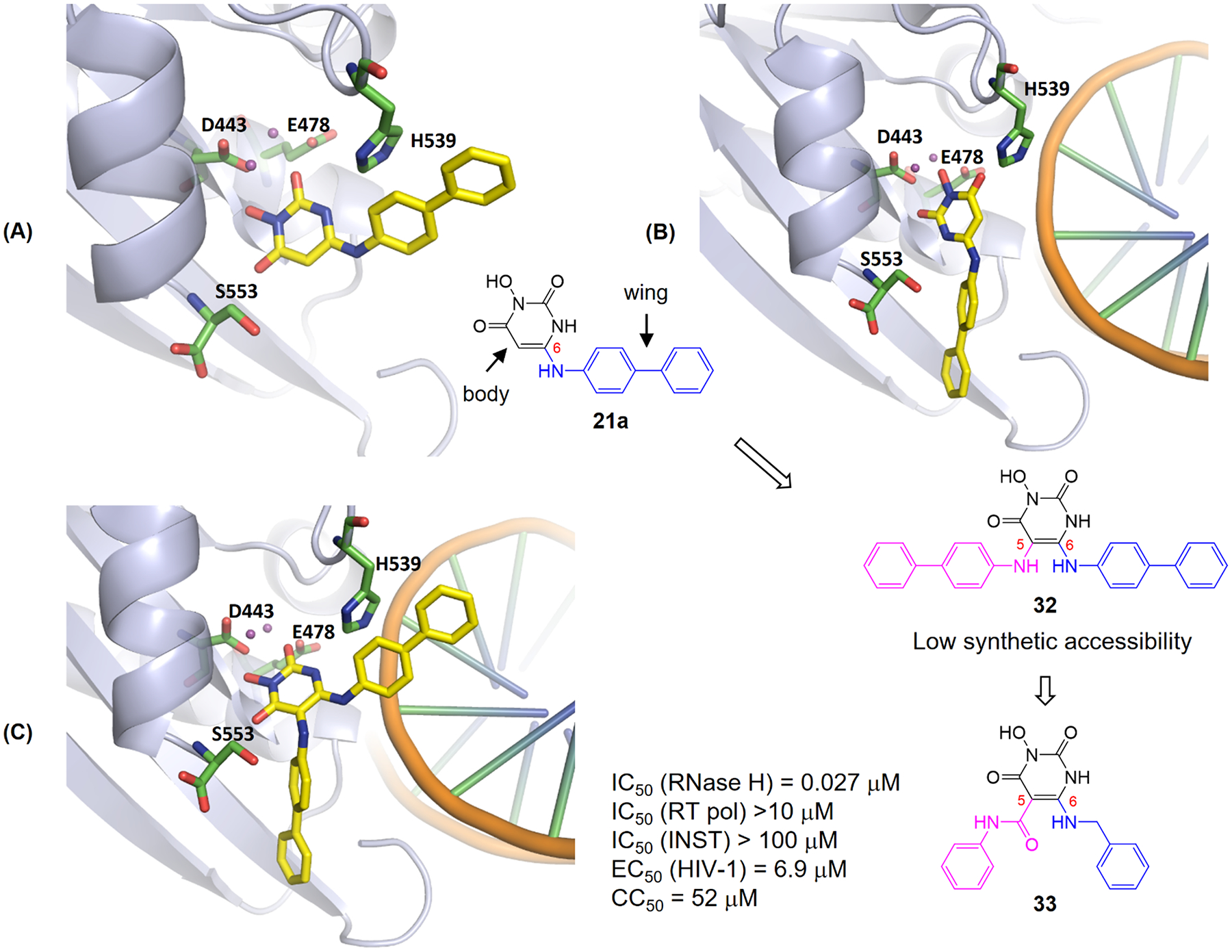
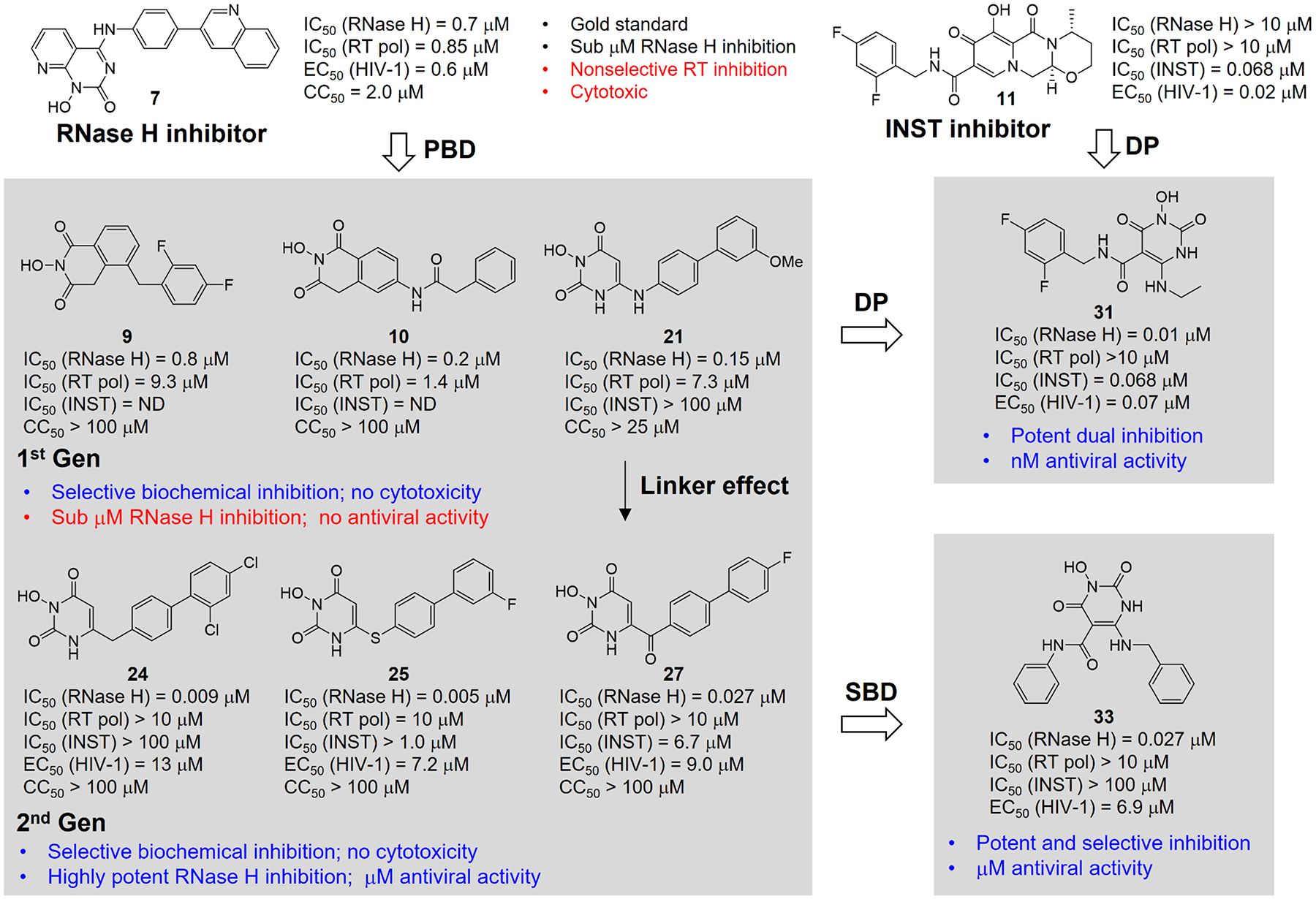
Human immunodeficiency virus (HIV) reverse transcriptase (RT) contains two distinct functional domains: a DNA polymerase (pol) domain and a ribonuclease H (RNase H) domain, both of which are required for viral genome replication. Over the last 3 decades, RT has been at the forefront of HIV drug discovery efforts with numerous nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs) approved by the FDA. However, all these RT inhibitors target only the pol function, and inhibitors of RT-associated RNase H have yet to enter the development pipeline, which in itself manifests both the opportunity and challenges of targeting RNase H: if developed, RT RNase H inhibitors would represent a mechanistically novel class of HIV drugs that can be particularly valuable in treating HIV strains resistant to current drugs. The challenges include (1) the difficulty in selectively targeting RT RNase H over RT pol due to their close interplay both spatially and temporally and over HIV-1 integrase strand transfer (INST) activity because of their active site similarities; (2) to a larger extent, the inability of active site inhibitors to confer significant antiviral effect, presumably due to a steep substrate barrier by which the pre-existing substrate prevents access of small molecules to the active site. As a result, previously reported RT RNase H inhibitors typically lacked target specificity and significant antiviral potency. Achieving meaningful antiviral activity via active site targeting likely entails selective and ultrapotent RNase H inhibition to allow small molecules to cut into the dominance of substrates. Based on a pharmacophore model informed by prior work, we designed and redesigned a few metal-chelating chemotypes, such as 2-hydroxyisoquinolinedione (HID), hydroxypyridonecarboxylic acid (HPCA), 3-hydroxypyrimidine-2,4-dione (HPD), and N-hydroxythienopyrimidine-2,4-dione (HTPD). Analogues of these chemotypes generally exhibited improved potency and selectivity inhibiting RT RNase H over the best previous compounds and further validated the pharmacophore model. Extended structure-activity relationship (SAR) on the HPD inhibitor type by mainly altering the linkage generated a few subtypes showing exceptional potency (single-digit nanomolar) and excellent selectivity over the inhibition of RT pol and INST. In parallel, a structure-based approach also allowed us to design a unique double-winged HPD subtype to potently and selectively inhibit RT RNase H and effectively compete against the RNA/DNA substrate. Significantly, all potent HPD subtypes consistently inhibited HIV-1 in the cell culture, suggesting that carefully designed active site RNase H inhibitors with ultrapotency could partially overcome the barrier to antiviral phenotype. Overall, in addition to identifying our own inhibitor types, our medicinal chemistry efforts demonstrated the value of pharmacophore and structure-based approaches in designing active side-directed RNase H inhibitors and could provide a viable path to validating RNase H as a novel antiviral target.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
Similar articles
-
6-Arylthio-3-hydroxypyrimidine-2,4-diones potently inhibited HIV reverse transcriptase-associated RNase H with antiviral activity.Eur J Med Chem. 2018 Aug 5;156:652-665. doi: 10.1016/j.ejmech.2018.07.039. Epub 2018 Jul 17. Eur J Med Chem. 2018. PMID: 30031976 Free PMC article.
-
Design, synthesis and biological evaluations of N-Hydroxy thienopyrimidine-2,4-diones as inhibitors of HIV reverse transcriptase-associated RNase H.Eur J Med Chem. 2017 Dec 1;141:149-161. doi: 10.1016/j.ejmech.2017.09.054. Epub 2017 Sep 28. Eur J Med Chem. 2017. PMID: 29031062 Free PMC article.
-
6-Cyclohexylmethyl-3-hydroxypyrimidine-2,4-dione as an inhibitor scaffold of HIV reverase transcriptase: Impacts of the 3-OH on inhibiting RNase H and polymerase.Eur J Med Chem. 2017 Mar 10;128:168-179. doi: 10.1016/j.ejmech.2017.01.041. Epub 2017 Jan 30. Eur J Med Chem. 2017. PMID: 28182989 Free PMC article.
-
Small-molecule Inhibitors of HIV-1 Reverse Transcriptase-Associated Ribonuclease H Function: Challenges and Recent Developments.Curr Med Chem. 2021;28(30):6146-6178. doi: 10.2174/0929867328666210322164557. Curr Med Chem. 2021. PMID: 34225606 Review.
-
Diketo acids derivatives as dual inhibitors of human immunodeficiency virus type 1 integrase and the reverse transcriptase RNase H domain.Curr Med Chem. 2011;18(22):3335-42. doi: 10.2174/092986711796504619. Curr Med Chem. 2011. PMID: 21728968 Review.
Cited by
-
4,5-Dihydroxypyrimidine Methyl Carboxylates, Carboxylic Acids, and Carboxamides as Inhibitors of Human Cytomegalovirus pUL89 Endonuclease.J Med Chem. 2022 Apr 14;65(7):5830-5849. doi: 10.1021/acs.jmedchem.2c00203. Epub 2022 Apr 4. J Med Chem. 2022. PMID: 35377638 Free PMC article.
-
Targeting Ribonucleases with Small Molecules and Bifunctional Molecules.ACS Chem Biol. 2023 Oct 20;18(10):2101-2113. doi: 10.1021/acschembio.3c00191. Epub 2023 Jun 29. ACS Chem Biol. 2023. PMID: 37382390 Free PMC article. Review.
-
Pyrroles as Privileged Scaffolds in the Search for New Potential HIV Inhibitors.Pharmaceuticals (Basel). 2021 Sep 2;14(9):893. doi: 10.3390/ph14090893. Pharmaceuticals (Basel). 2021. PMID: 34577593 Free PMC article. Review.
-
Repurposing N-hydroxy thienopyrimidine-2,4-diones (HtPD) as inhibitors of human cytomegalovirus pUL89 endonuclease: Synthesis and biological characterization.Bioorg Chem. 2022 Dec;129:106198. doi: 10.1016/j.bioorg.2022.106198. Epub 2022 Oct 12. Bioorg Chem. 2022. PMID: 36265353 Free PMC article.
-
Avoiding Drug Resistance in HIV Reverse Transcriptase.Chem Rev. 2021 Mar 24;121(6):3271-3296. doi: 10.1021/acs.chemrev.0c00967. Epub 2021 Jan 28. Chem Rev. 2021. PMID: 33507067 Free PMC article. Review.
References
-
- Hirsch MS AIDS Commentary. Azidothymidine. The Journal of Infectious Diseases, 1988, 157, 427–431. - PubMed
-
- U.S. Food and Drug Administration, Antiretroviral drugs used in the treatment of HIV infection, April 12, 2018. www.fda.gov.
-
- Yeni P Update on HAART in HIV. J. Hepatol 2006, 44, S100–103. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
