

SpliceFinder: ab initio prediction of splice sites using convolutional neural network
- PMID: 31881982
- PMCID: PMC6933889
- DOI: 10.1186/s12859-019-3306-3
SpliceFinder: ab initio prediction of splice sites using convolutional neural network
Abstract
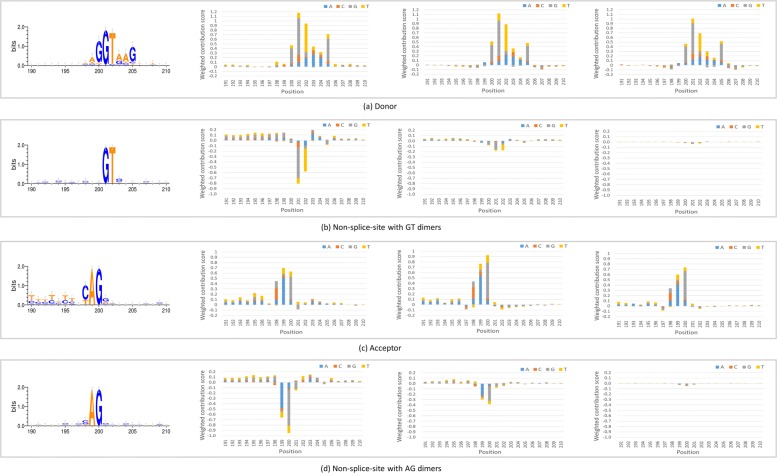
Background: Identifying splice sites is a necessary step to analyze the location and structure of genes. Two dinucleotides, GT and AG, are highly frequent on splice sites, and many other patterns are also on splice sites with important biological functions. Meanwhile, the dinucleotides occur frequently at the sequences without splice sites, which makes the prediction prone to generate false positives. Most existing tools select all the sequences with the two dimers and then focus on distinguishing the true splice sites from those pseudo ones. Such an approach will lead to a decrease in false positives; however, it will result in non-canonical splice sites missing.
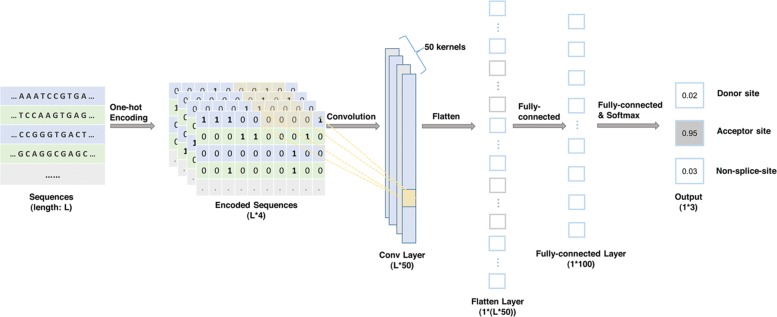
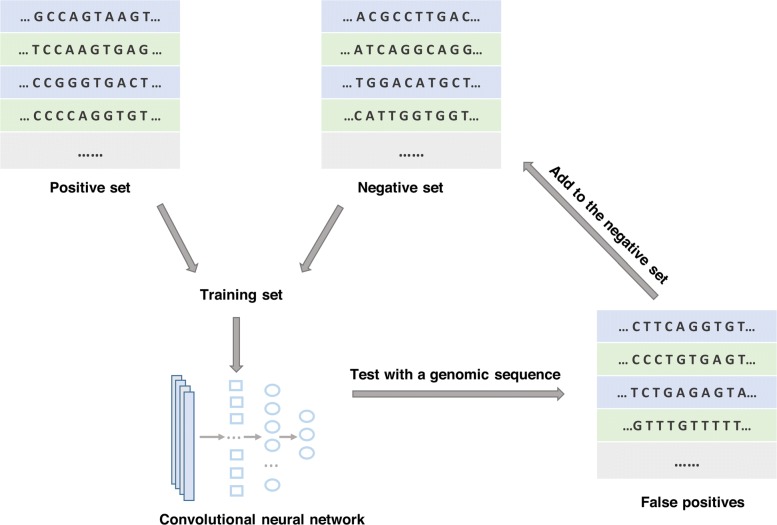
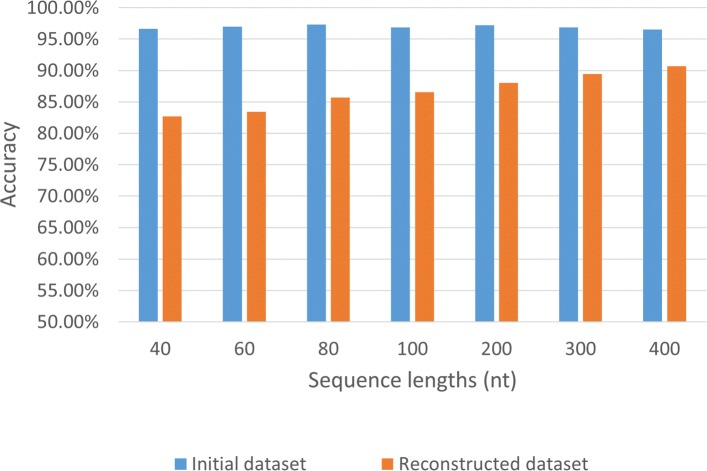
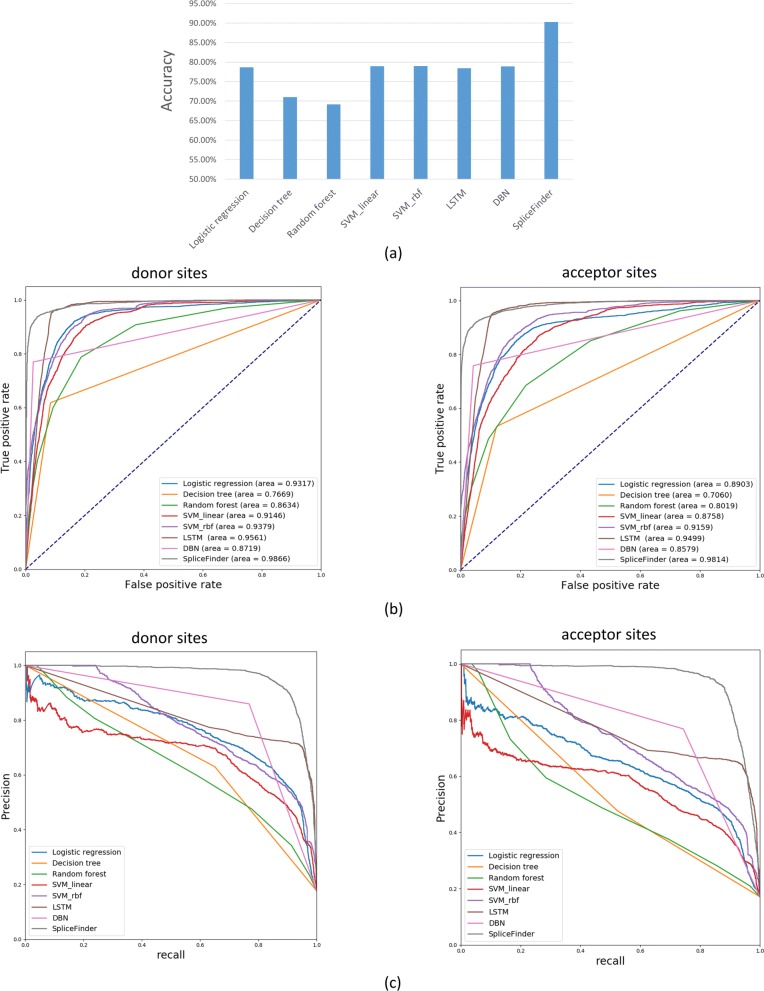
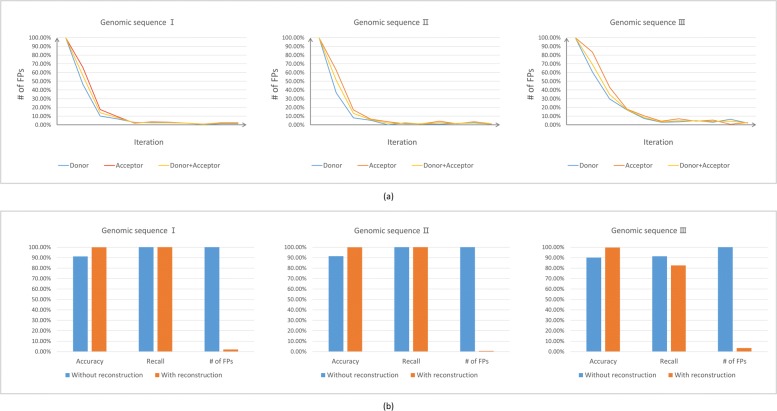
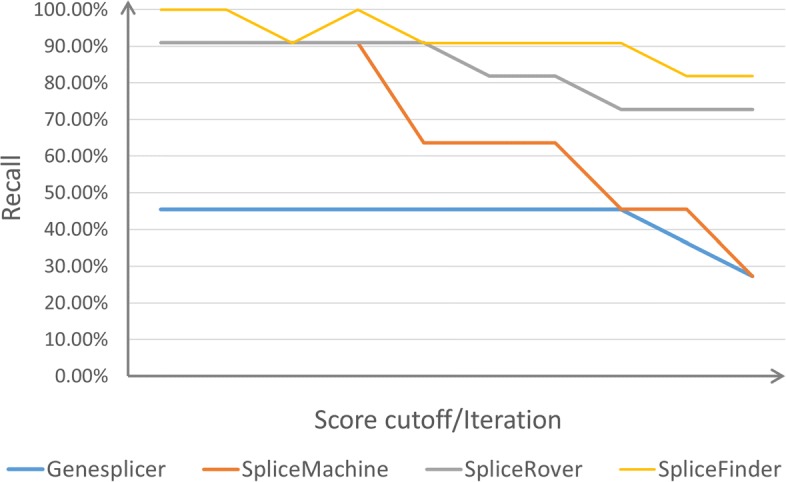
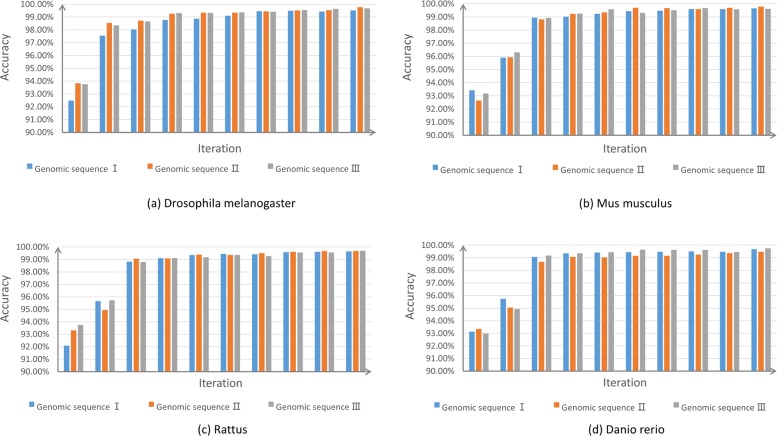
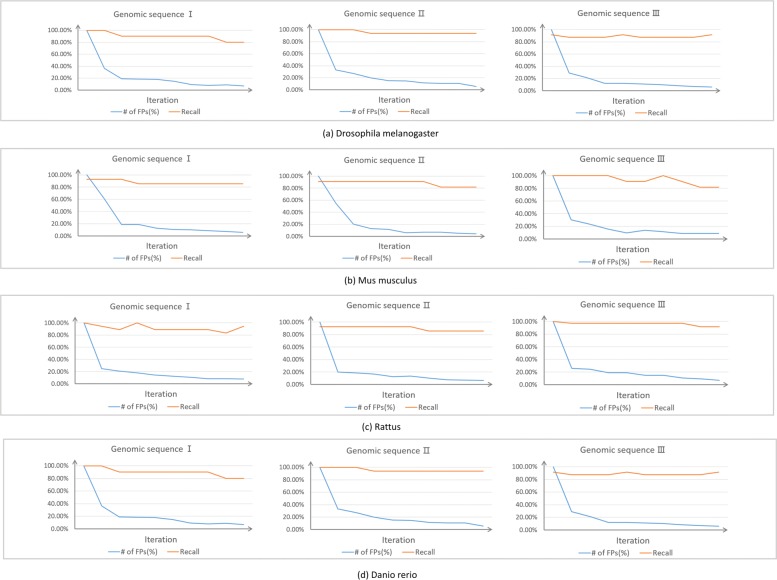
Result: We have designed SpliceFinder based on convolutional neural network (CNN) to predict splice sites. To achieve the ab initio prediction, we used human genomic data to train our neural network. An iterative approach is adopted to reconstruct the dataset, which tackles the data unbalance problem and forces the model to learn more features of splice sites. The proposed CNN obtains the classification accuracy of 90.25%, which is 10% higher than the existing algorithms. The method outperforms other existing methods in terms of area under receiver operating characteristics (AUC), recall, precision, and F1 score. Furthermore, SpliceFinder can find the exact position of splice sites on long genomic sequences with a sliding window. Compared with other state-of-the-art splice site prediction tools, SpliceFinder generates results in about half lower false positive while keeping recall higher than 0.8. Also, SpliceFinder captures the non-canonical splice sites. In addition, SpliceFinder performs well on the genomic sequences of Drosophila melanogaster, Mus musculus, Rattus, and Danio rerio without retraining.
Conclusion: Based on CNN, we have proposed a new ab initio splice site prediction tool, SpliceFinder, which generates less false positives and can detect non-canonical splice sites. Additionally, SpliceFinder is transferable to other species without retraining. The source code and additional materials are available at https://gitlab.deepomics.org/wangruohan/SpliceFinder.
Keywords: Canonical and non-canonical splice sites; Convolutional neural network; Splice site prediction.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Analysis of canonical and non-canonical splice sites in mammalian genomes.Nucleic Acids Res. 2000 Nov 1;28(21):4364-75. doi: 10.1093/nar/28.21.4364. Nucleic Acids Res. 2000. PMID: 11058137 Free PMC article.
-
Human Splice-Site Prediction with Deep Neural Networks.J Comput Biol. 2018 Aug;25(8):954-961. doi: 10.1089/cmb.2018.0041. Epub 2018 Apr 18. J Comput Biol. 2018. PMID: 29668310
-
Read-Split-Run: an improved bioinformatics pipeline for identification of genome-wide non-canonical spliced regions using RNA-Seq data.BMC Genomics. 2016 Aug 22;17 Suppl 7(Suppl 7):503. doi: 10.1186/s12864-016-2896-7. BMC Genomics. 2016. PMID: 27556805 Free PMC article.
-
Comprehensive review and assessment of computational methods for predicting RNA post-transcriptional modification sites from RNA sequences.Brief Bioinform. 2020 Sep 25;21(5):1676-1696. doi: 10.1093/bib/bbz112. Brief Bioinform. 2020. PMID: 31714956 Review.
-
Convolutional Neural Networks for ATC Classification.Curr Pharm Des. 2018;24(34):4007-4012. doi: 10.2174/1381612824666181112113438. Curr Pharm Des. 2018. PMID: 30417778 Review.
Cited by
-
Principles and Practical Considerations for the Analysis of Disease-Associated Alternative Splicing Events Using the Gateway Cloning-Based Minigene Vectors pDESTsplice and pSpliceExpress.Int J Mol Sci. 2021 May 13;22(10):5154. doi: 10.3390/ijms22105154. Int J Mol Sci. 2021. PMID: 34068052 Free PMC article. Review.
-
Improved recognition of splice sites in A. thaliana by incorporating secondary structure information into sequence-derived features: a computational study.3 Biotech. 2021 Nov;11(11):484. doi: 10.1007/s13205-021-03036-8. Epub 2021 Oct 31. 3 Biotech. 2021. PMID: 34790508 Free PMC article.
-
Splam: a deep-learning-based splice site predictor that improves spliced alignments.Genome Biol. 2024 Sep 16;25(1):243. doi: 10.1186/s13059-024-03379-4. Genome Biol. 2024. PMID: 39285451 Free PMC article.
-
Splice Junction Identification using Long Short-Term Memory Neural Networks.Curr Genomics. 2021 Dec 30;22(5):384-390. doi: 10.2174/1389202922666211011143008. Curr Genomics. 2021. PMID: 35283668 Free PMC article.
-
Applications for Deep Learning in Epilepsy Genetic Research.Int J Mol Sci. 2023 Sep 27;24(19):14645. doi: 10.3390/ijms241914645. Int J Mol Sci. 2023. PMID: 37834093 Free PMC article. Review.
References
-
- Reese MG, Eeckman FH, Kulp D, Haussler D. Improved splice site detection in genie. J Comput Biol. 1997;4(3):311–23. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
