

Interleukin-1β drives NEDD8 nuclear-to-cytoplasmic translocation, fostering parkin activation via NEDD8 binding to the P-ubiquitin activating site
- PMID: 31882005
- PMCID: PMC6935243
- DOI: 10.1186/s12974-019-1669-z
Interleukin-1β drives NEDD8 nuclear-to-cytoplasmic translocation, fostering parkin activation via NEDD8 binding to the P-ubiquitin activating site
Abstract
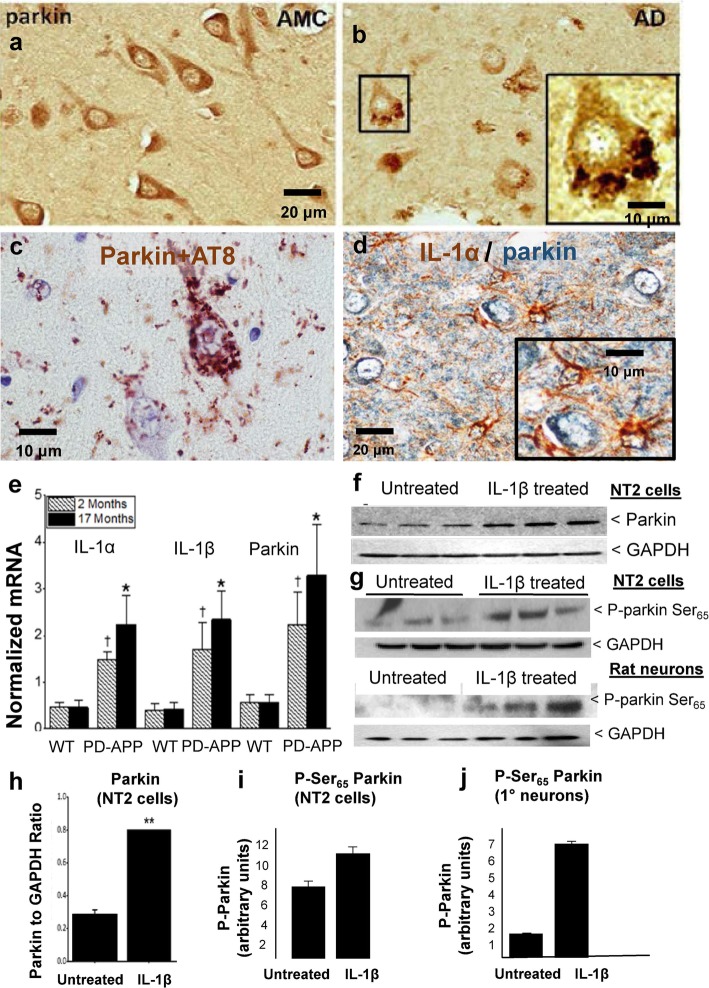
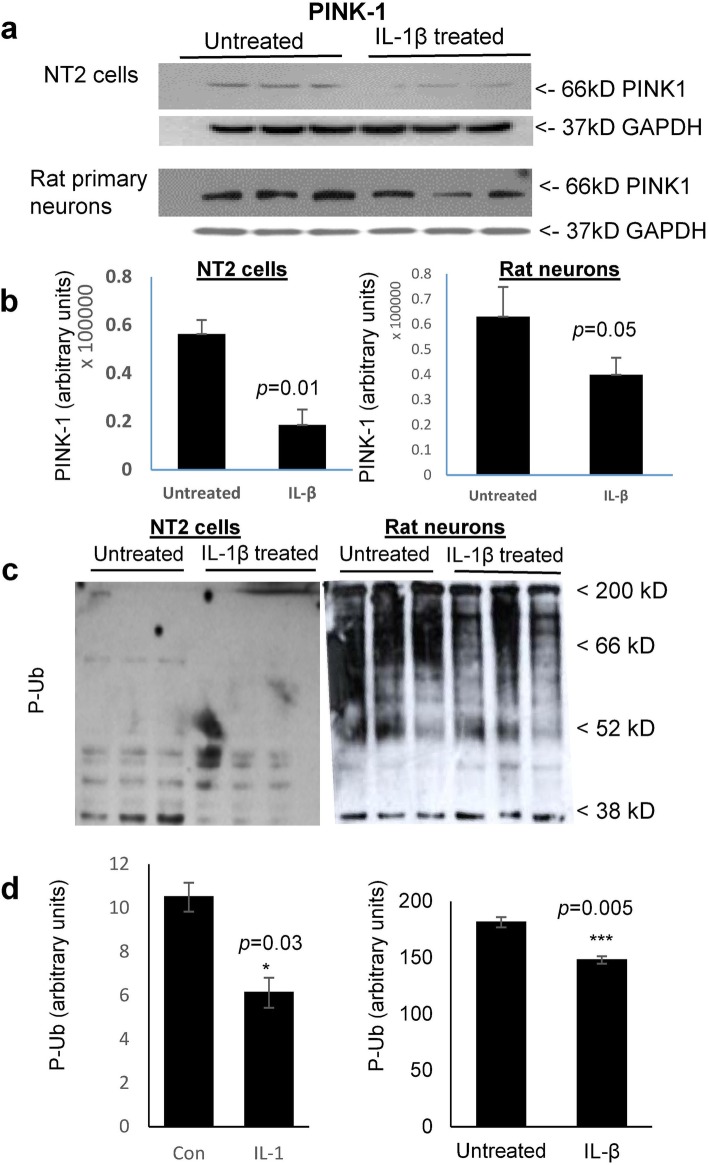
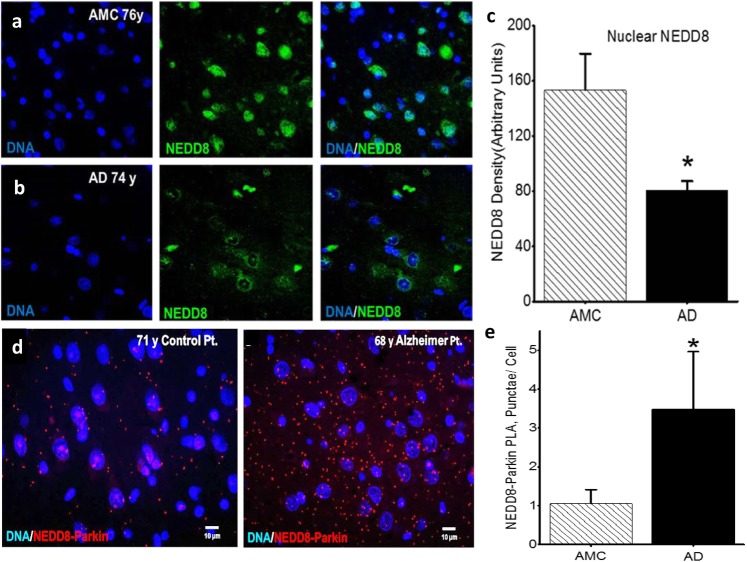
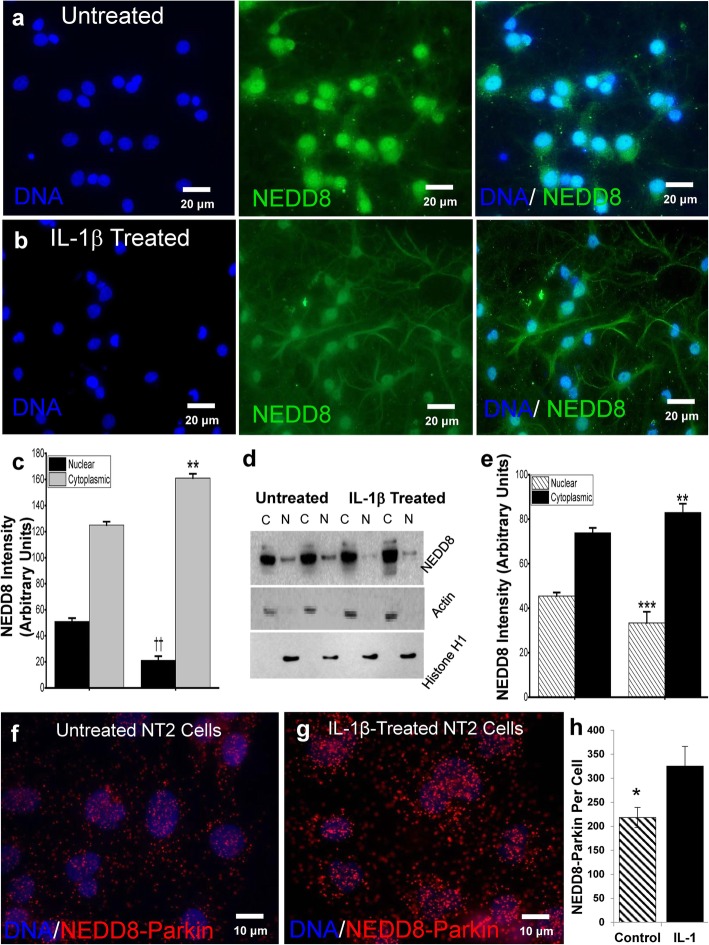
Background: Neuroinflammation, typified by elevated levels of interleukin-1 (IL-1) α and β, and deficits in proteostasis, characterized by accumulation of polyubiquitinated proteins and other aggregates, are associated with neurodegenerative disease independently and through interactions of the two phenomena. We investigated the influence of IL-1β on ubiquitination via its impact on activation of the E3 ligase parkin by either phosphorylated ubiquitin (P-Ub) or NEDD8.
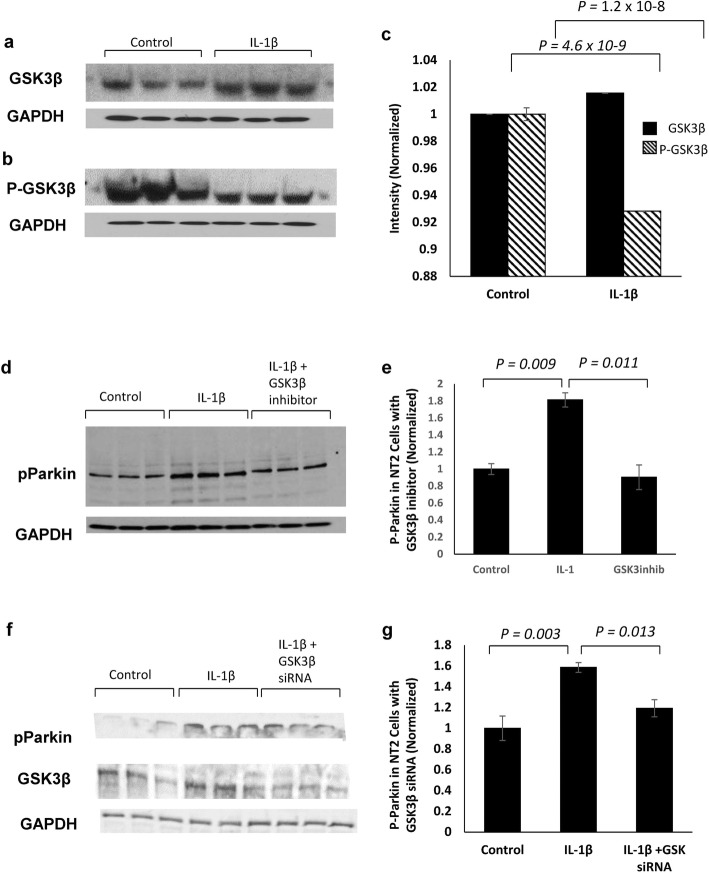
Methods: Immunohistochemistry and Proximity Ligation Assay were used to assess colocalization of parkin with P-tau or NEDD8 in hippocampus from Alzheimer patients (AD) and controls. IL-1β effects on PINK1, P-Ub, parkin, P-parkin, and GSK3β-as well as phosphorylation of parkin by GSK3β-were assessed in cell cultures by western immunoblot, using two inhibitors and siRNA knockdown to suppress GSK3β. Computer modeling characterized the binding and the effects of P-Ub and NEDD8 on parkin. IL-1α, IL-1β, and parkin gene expression was assessed by RT-PCR in brains of 2- and 17-month-old PD-APP mice and wild-type littermates.
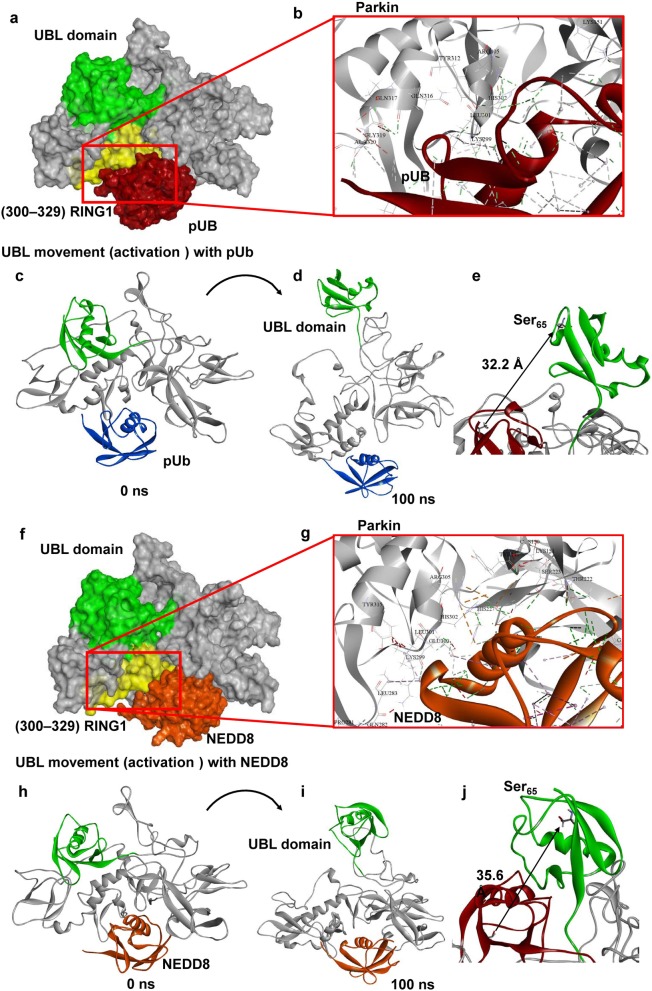
Results: IL-1α, IL-1β, and parkin mRNA levels were higher in PD-APP mice compared with wild-type littermates, and IL-1α-laden glia surrounded parkin- and P-tau-laden neurons in human AD. Such neurons showed a nuclear-to-cytoplasmic translocation of NEDD8 that was mimicked in IL-1β-treated primary neuronal cultures. These cultures also showed higher parkin levels and GSK3β-induced parkin phosphorylation; PINK1 levels were suppressed. In silico simulation predicted that binding of either P-Ub or NEDD8 at a singular position on parkin opens the UBL domain, exposing Ser65 for parkin activation.
Conclusions: The promotion of parkin- and NEDD8-mediated ubiquitination by IL-1β is consistent with an acute neuroprotective role. However, accumulations of P-tau and P-Ub and other elements of proteostasis, such as translocated NEDD8, in AD and in response to IL-1β suggest either over-stimulation or a proteostatic failure that may result from chronic IL-1β elevation, easily envisioned considering its early induction in Down's syndrome and mild cognitive impairment. The findings further link autophagy and neuroinflammation, two important aspects of AD pathogenesis, which have previously been only loosely related.
Keywords: Alzheimer’s; Autophagy; GSK3β; IL-1β; NEDD8; Neddylation; PINK1; Parkin; Simulation interactions; Ubiquitination.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
