

Multi-species annotation of transcriptome and chromatin structure in domesticated animals
- PMID: 31884969
- PMCID: PMC6936065
- DOI: 10.1186/s12915-019-0726-5
Multi-species annotation of transcriptome and chromatin structure in domesticated animals
Abstract
Background: Comparative genomics studies are central in identifying the coding and non-coding elements associated with complex traits, and the functional annotation of genomes is a critical step to decipher the genotype-to-phenotype relationships in livestock animals. As part of the Functional Annotation of Animal Genomes (FAANG) action, the FR-AgENCODE project aimed to create reference functional maps of domesticated animals by profiling the landscape of transcription (RNA-seq), chromatin accessibility (ATAC-seq) and conformation (Hi-C) in species representing ruminants (cattle, goat), monogastrics (pig) and birds (chicken), using three target samples related to metabolism (liver) and immunity (CD4+ and CD8+ T cells).
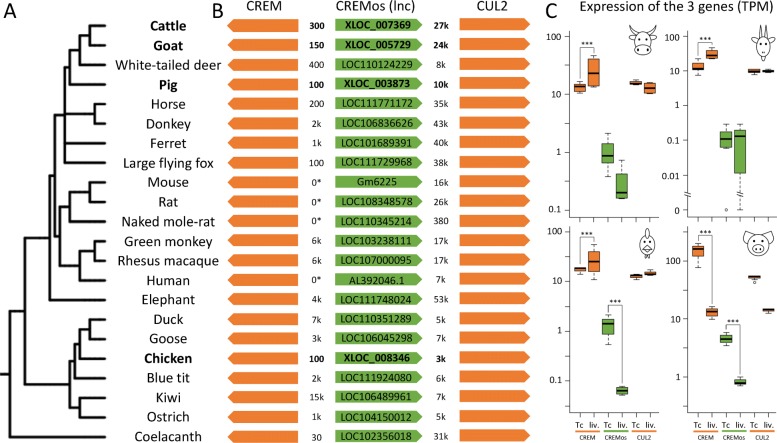
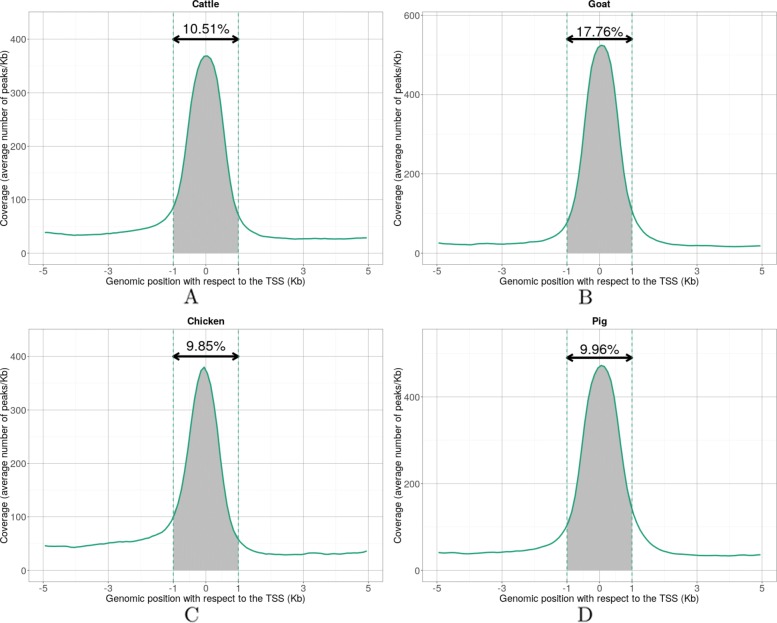
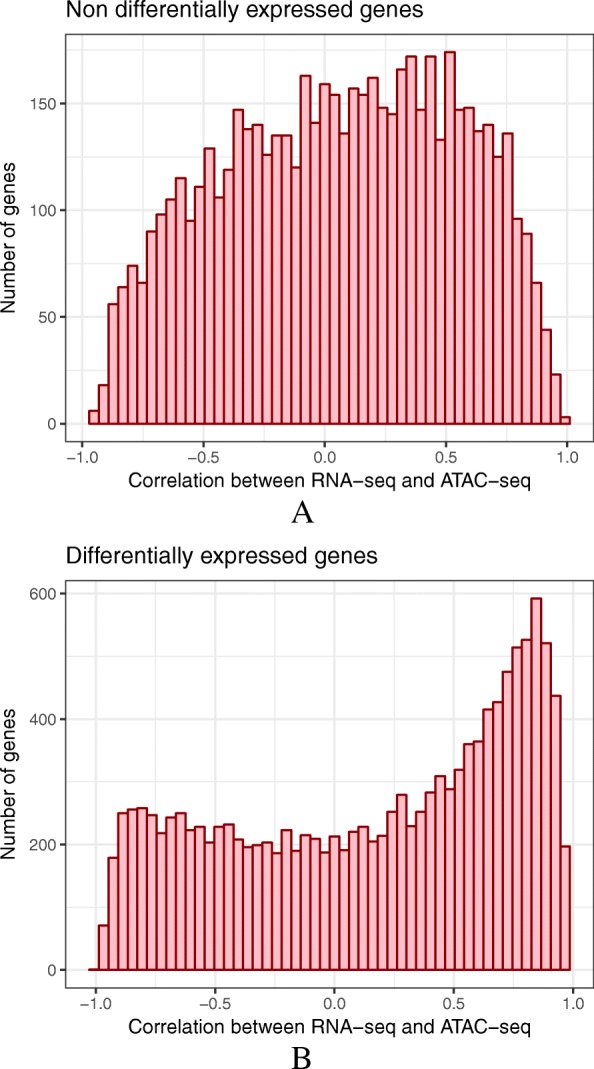
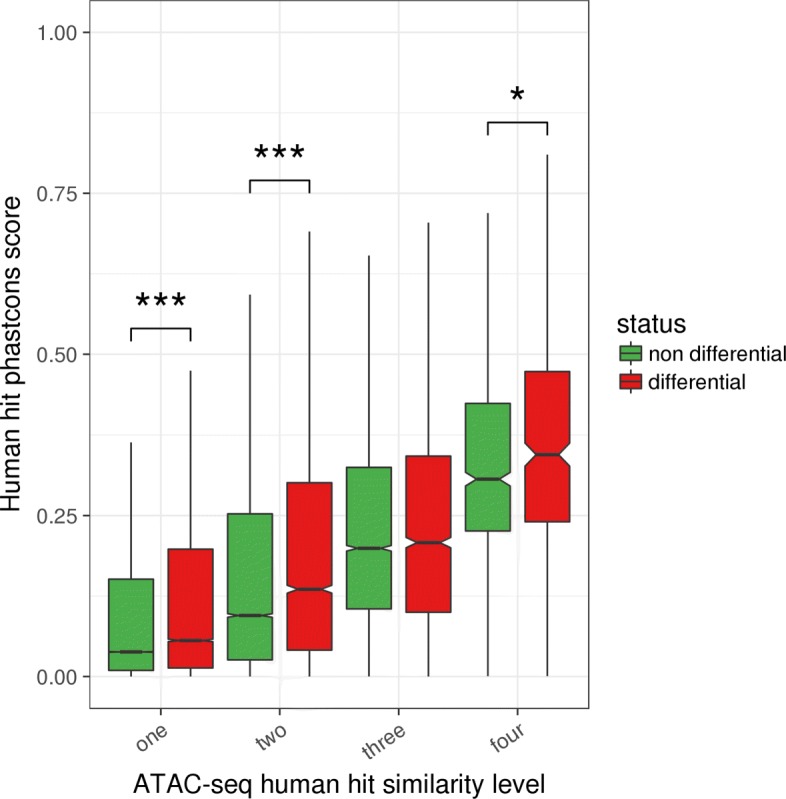
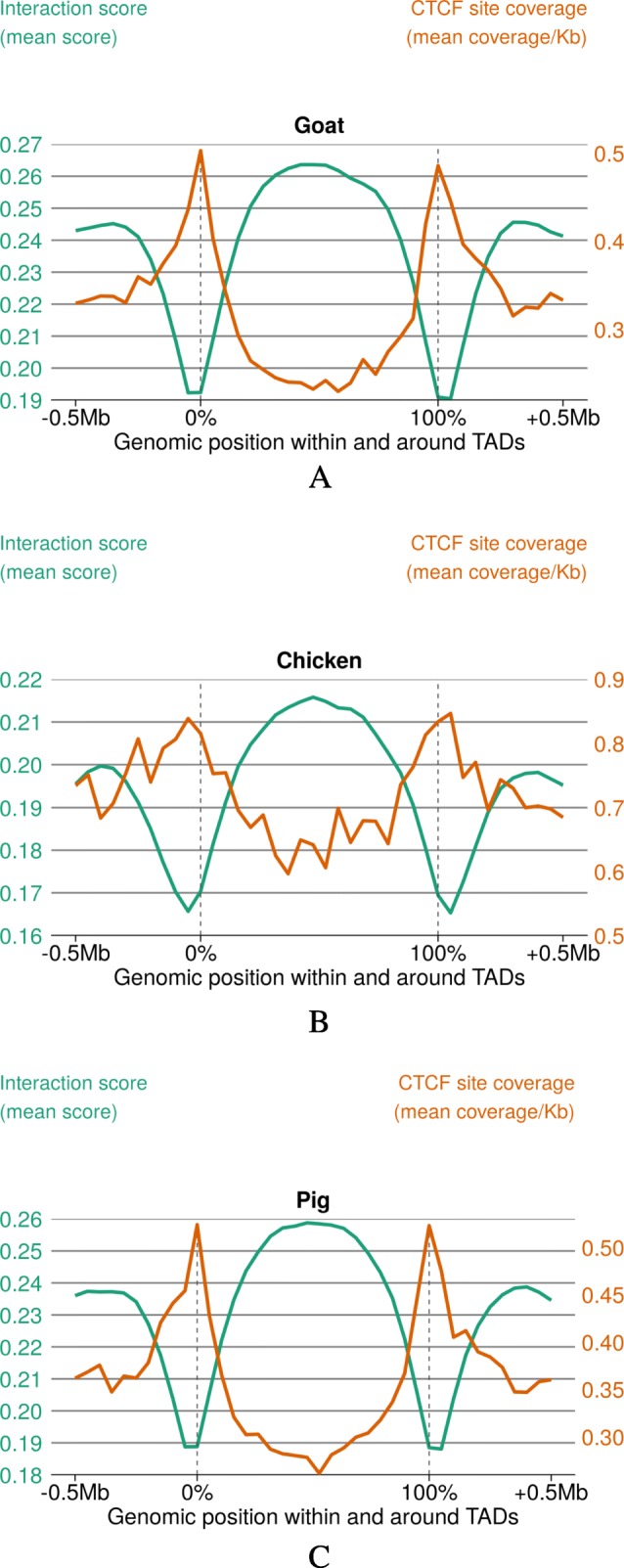
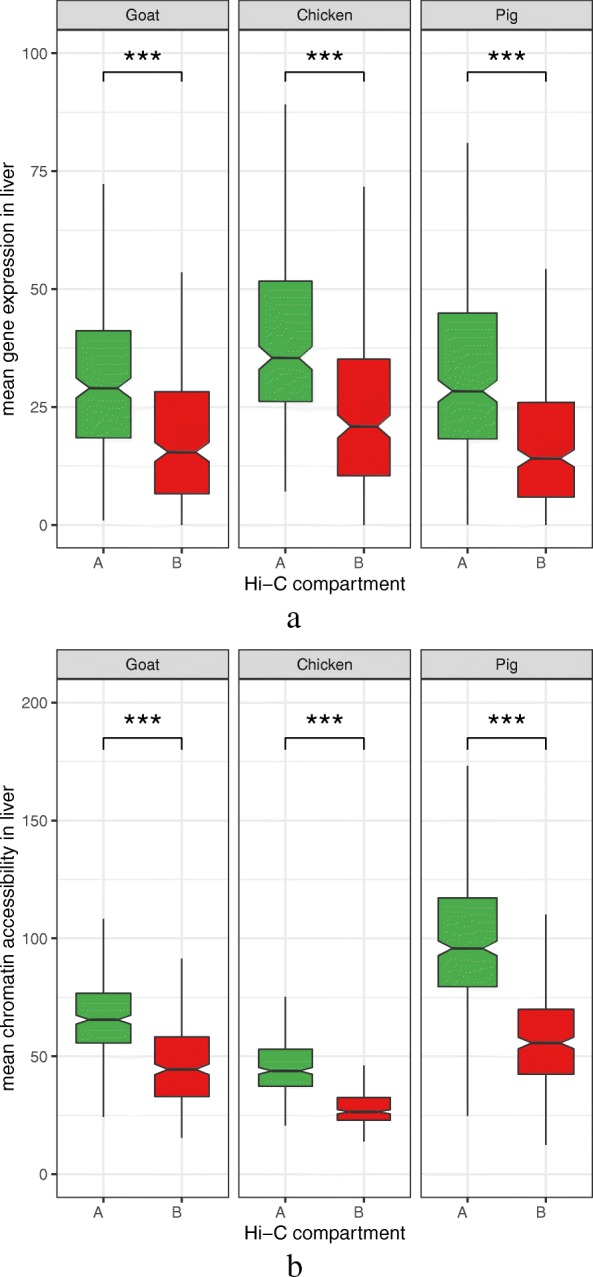
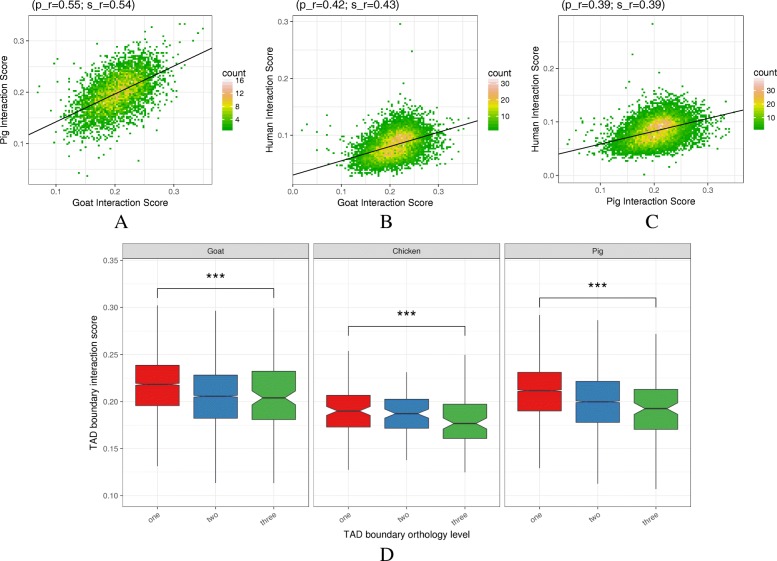
Results: RNA-seq assays considerably extended the available catalog of annotated transcripts and identified differentially expressed genes with unknown function, including new syntenic lncRNAs. ATAC-seq highlighted an enrichment for transcription factor binding sites in differentially accessible regions of the chromatin. Comparative analyses revealed a core set of conserved regulatory regions across species. Topologically associating domains (TADs) and epigenetic A/B compartments annotated from Hi-C data were consistent with RNA-seq and ATAC-seq data. Multi-species comparisons showed that conserved TAD boundaries had stronger insulation properties than species-specific ones and that the genomic distribution of orthologous genes in A/B compartments was significantly conserved across species.
Conclusions: We report the first multi-species and multi-assay genome annotation results obtained by a FAANG project. Beyond the generation of reference annotations and the confirmation of previous findings on model animals, the integrative analysis of data from multiple assays and species sheds a new light on the multi-scale selective pressure shaping genome organization from birds to mammals. Overall, these results emphasize the value of FAANG for research on domesticated animals and reinforces the importance of future meta-analyses of the reference datasets being generated by this community on different species.
Keywords: ATAC-seq; Functional annotation; Hi-C; Livestock; RNA-seq.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
