

The Role of Cyclic AMP Signaling in Cardiac Fibrosis
- PMID: 31888098
- PMCID: PMC7016856
- DOI: 10.3390/cells9010069
The Role of Cyclic AMP Signaling in Cardiac Fibrosis
Abstract
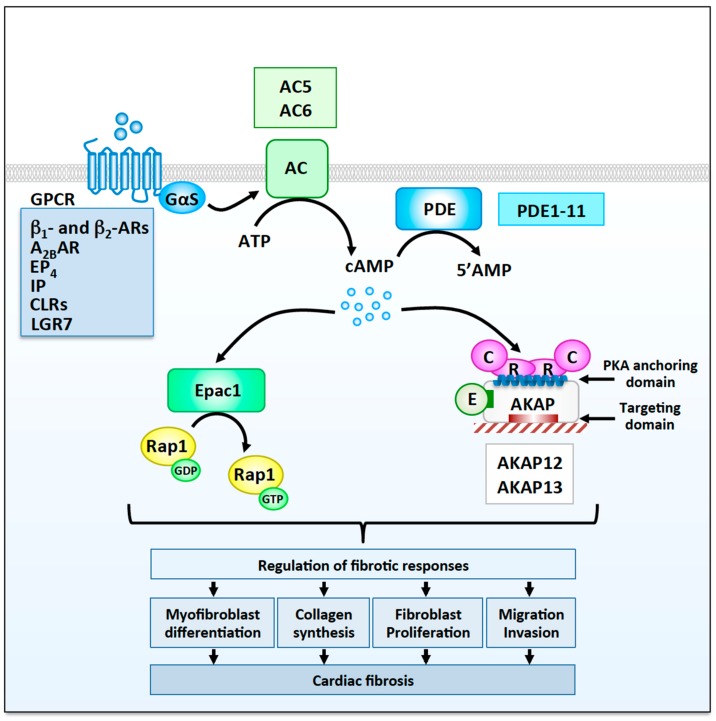
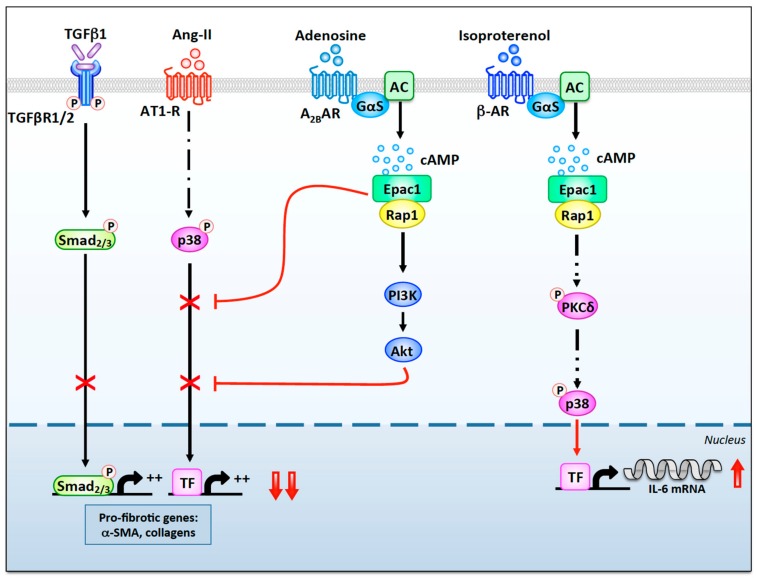
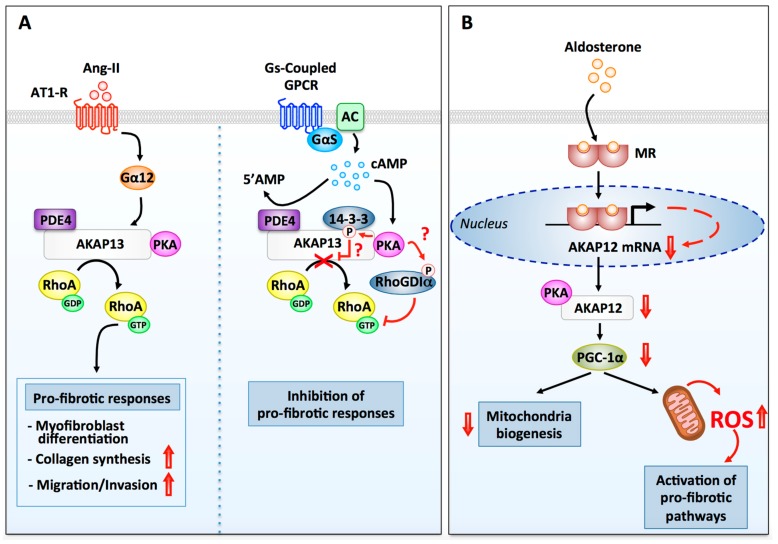
Myocardial stress and injury invariably promote remodeling of the cardiac tissue, which is associated with cardiomyocyte death and development of fibrosis. The fibrotic process is initially triggered by the differentiation of resident cardiac fibroblasts into myofibroblasts. These activated fibroblasts display increased proliferative capacity and secrete large amounts of extracellular matrix. Uncontrolled myofibroblast activation can thus promote heart stiffness, cardiac dysfunction, arrhythmias, and progression to heart failure. Despite the well-established role of myofibroblasts in mediating cardiac disease, our current knowledge on how signaling pathways promoting fibrosis are regulated and coordinated in this cell type is largely incomplete. In this respect, cyclic adenosine monophosphate (cAMP) signaling acts as a major modulator of fibrotic responses activated in fibroblasts of injured or stressed hearts. In particular, accumulating evidence now suggests that upstream cAMP modulators including G protein-coupled receptors, adenylyl cyclases (ACs), and phosphodiesterases (PDEs); downstream cAMP effectors such as protein kinase A (PKA) and the guanine nucleotide exchange factor Epac; and cAMP signaling organizers such as A-kinase anchoring proteins (AKAPs) modulate a variety of fundamental cellular processes involved in myocardial fibrosis including myofibroblast differentiation, proliferation, collagen secretion, and invasiveness. The current review will discuss recent advances highlighting the role of cAMP and AKAP-mediated signaling in regulating pathophysiological responses controlling cardiac fibrosis.
Keywords: A-kinase anchoring protein (AKAP), adenylyl cyclase; cardiac fibrosis; cardiac remodeling; cyclic AMP; phosphodiesterase; protein kinase A.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
