

Variants in Miro1 Cause Alterations of ER-Mitochondria Contact Sites in Fibroblasts from Parkinson's Disease Patients
- PMID: 31888276
- PMCID: PMC6947516
- DOI: 10.3390/jcm8122226
Variants in Miro1 Cause Alterations of ER-Mitochondria Contact Sites in Fibroblasts from Parkinson's Disease Patients
Abstract
Background: Although most cases of Parkinson´s disease (PD) are idiopathic with unknown cause, an increasing number of genes and genetic risk factors have been discovered that play a role in PD pathogenesis. Many of the PD-associated proteins are involved in mitochondrial quality control, e.g., PINK1, Parkin, and LRRK2, which were recently identified as regulators of mitochondrial-endoplasmic reticulum (ER) contact sites (MERCs) linking mitochondrial homeostasis to intracellular calcium handling. In this context, Miro1 is increasingly recognized to play a role in PD pathology. Recently, we identified the first PD patients carrying mutations in RHOT1, the gene coding for Miro1. Here, we describe two novel RHOT1 mutations identified in two PD patients and the characterization of the cellular phenotypes.
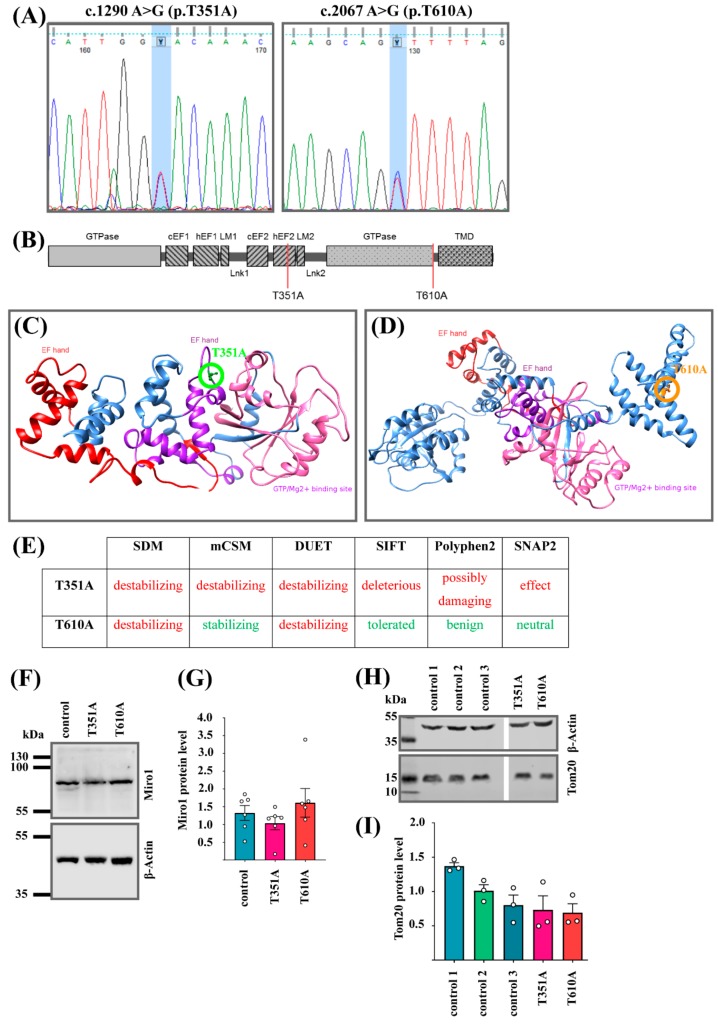
Methods: Using whole exome sequencing we identified two PD patients carrying heterozygous mutations leading to the amino acid exchanges T351A and T610A in Miro1. We analyzed calcium homeostasis and MERCs in detail by live cell imaging and immunocytochemistry in patient-derived fibroblasts.
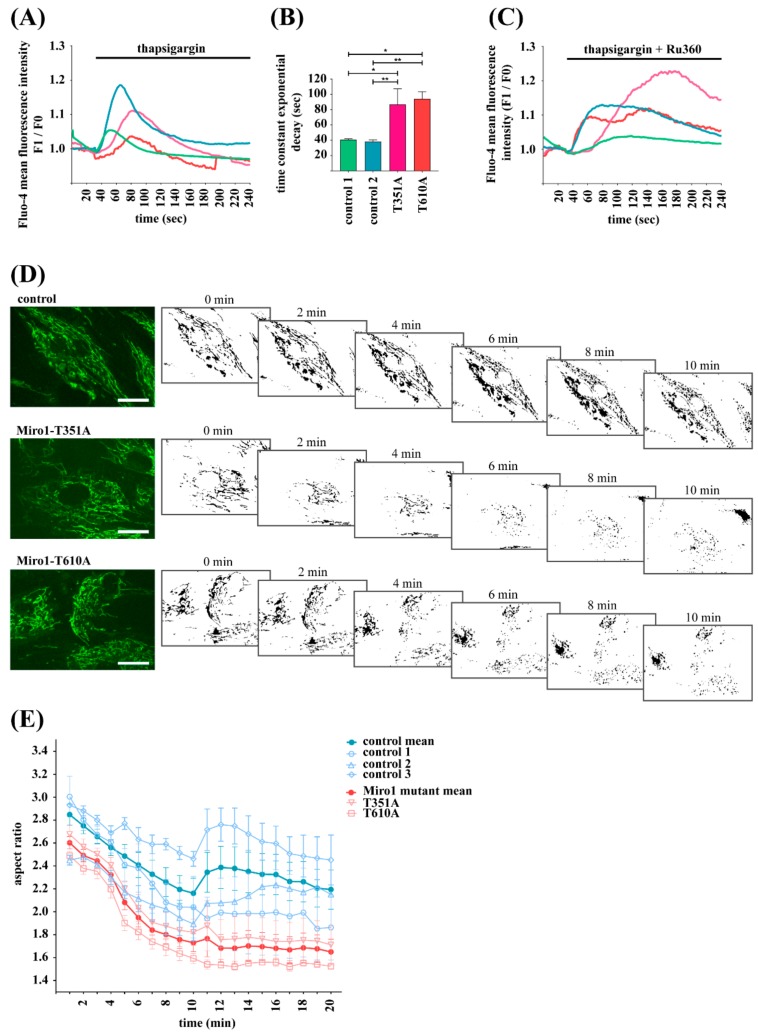
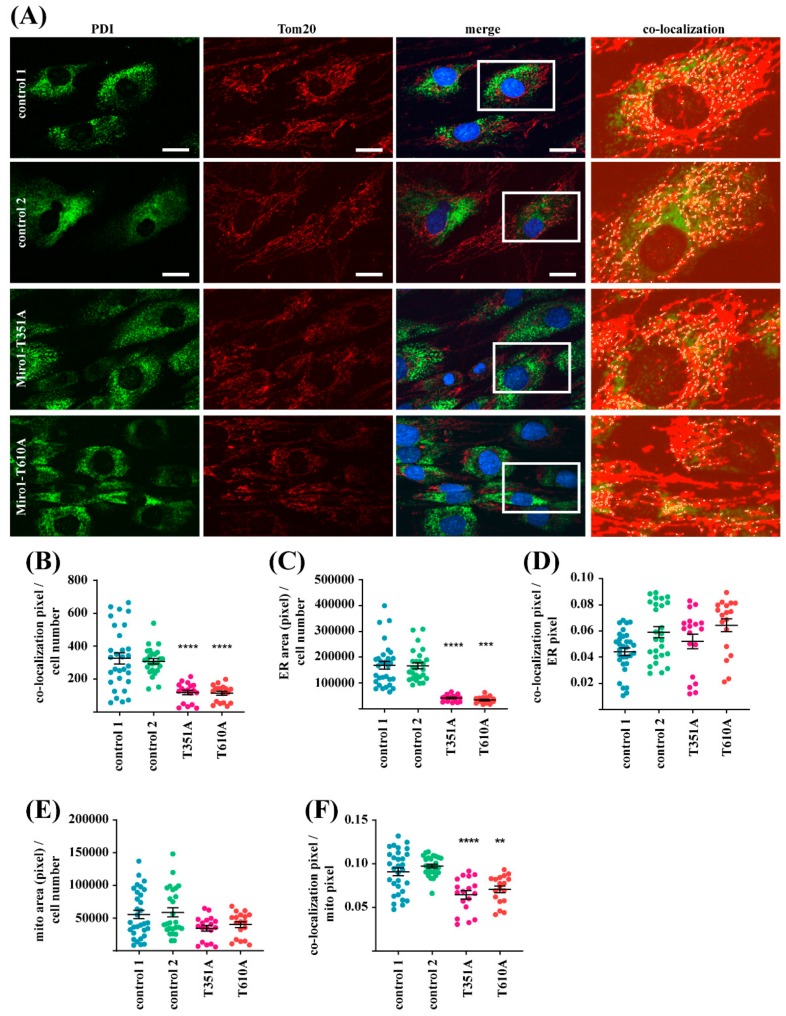
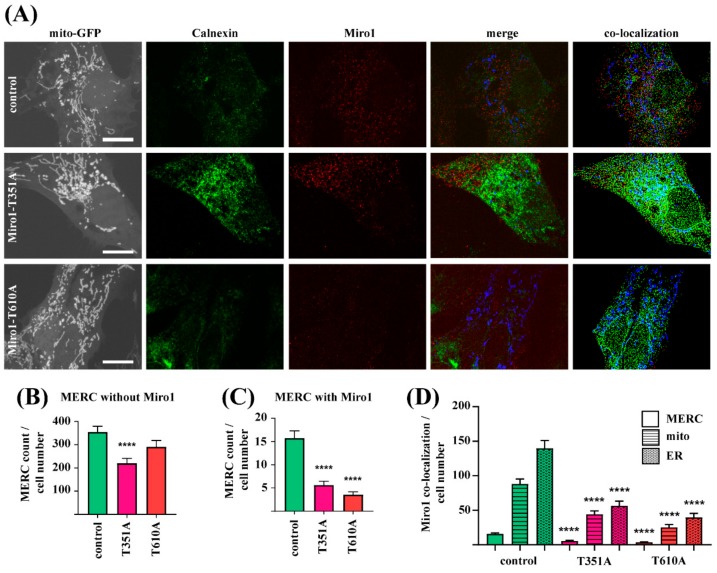
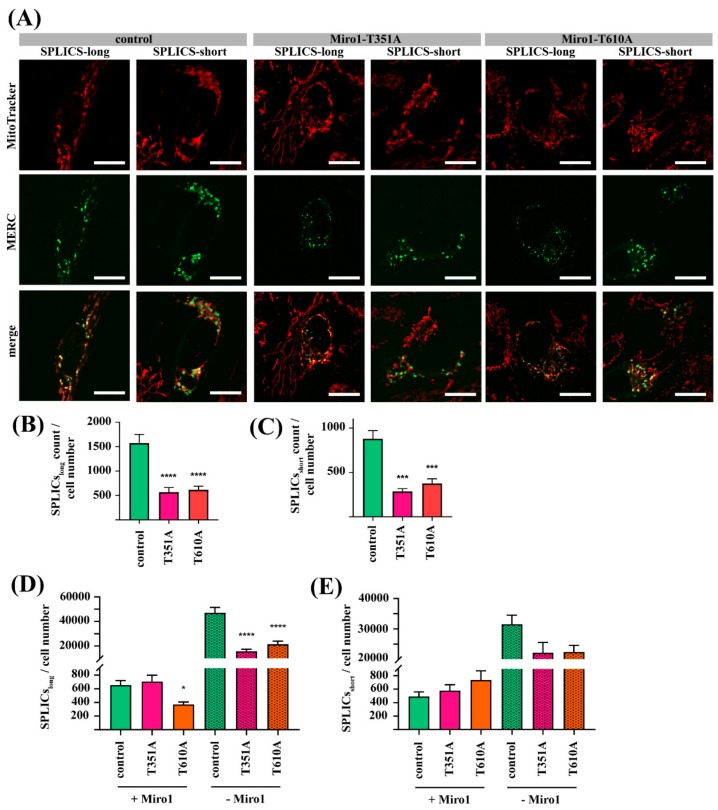
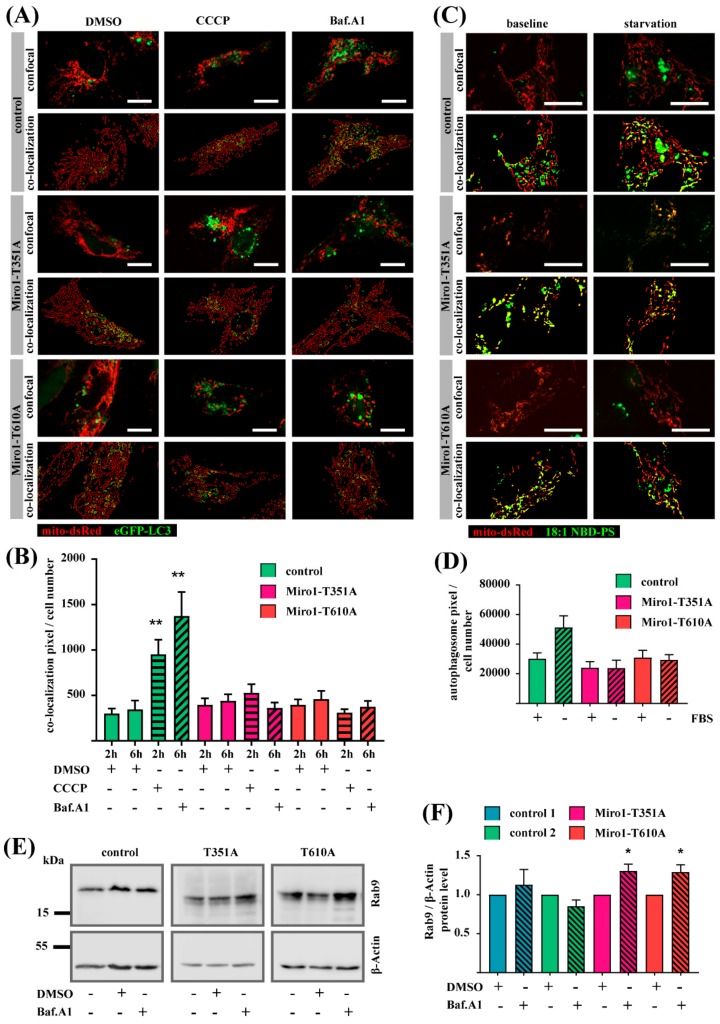
Results: We show that fibroblasts expressing mutant T351A or T610A Miro1 display impaired calcium homeostasis and a reduced amount of MERCs. All fibroblast lines from patients with pathogenic variants in Miro1, revealed alterations of the structure of MERCs.
Conclusion: Our data suggest that Miro1 is important for the regulation of the structure and function of MERCs. Moreover, our study supports the role of MERCs in the pathogenesis of PD and further establishes variants in RHOT1 as rare genetic risk factors for neurodegeneration.
Keywords: Miro1; Parkinson´s disease; mitochondria-ER contact sites.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Hsieh C.-H., Shaltouki A., Gonzalez A.E., Bettencourt da Cruz A., Burbulla L.F., St Lawrence E., Schüle B., Krainc D., Palmer T.D., Wang X. Functional Impairment in Miro Degradation and Mitophagy Is a Shared Feature in Familial and Sporadic Parkinson’s Disease. Cell Stem Cell. 2016;19:709–724. doi: 10.1016/j.stem.2016.08.002. - DOI - PMC - PubMed
-
- Lee K.-S., Huh S., Lee S., Wu Z., Kim A.-K., Kang H.-Y., Lu B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA. 2018;115:E8844–E8853. doi: 10.1073/pnas.1721136115. - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
