

Disorders of FZ-CRD; insights towards FZ-CRD folding and therapeutic landscape
- PMID: 31892318
- PMCID: PMC6938638
- DOI: 10.1186/s10020-019-0129-7
Disorders of FZ-CRD; insights towards FZ-CRD folding and therapeutic landscape
Abstract
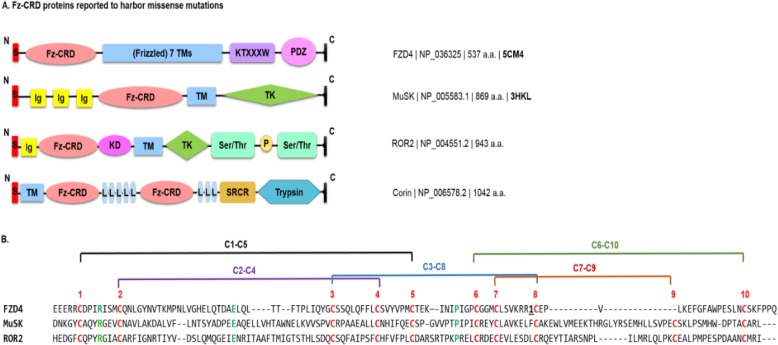
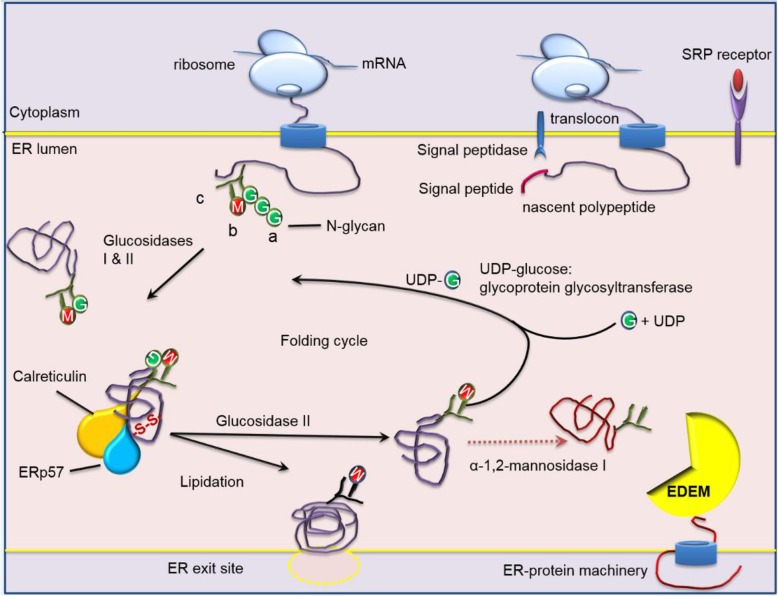
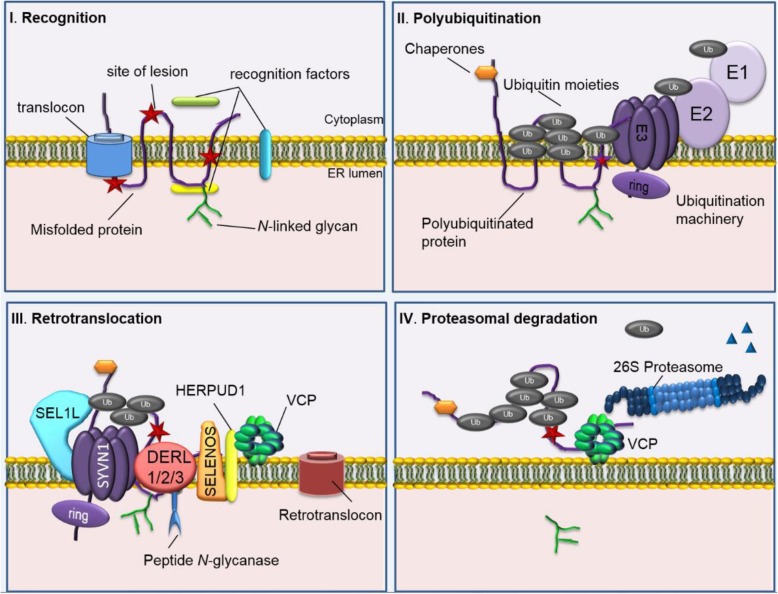
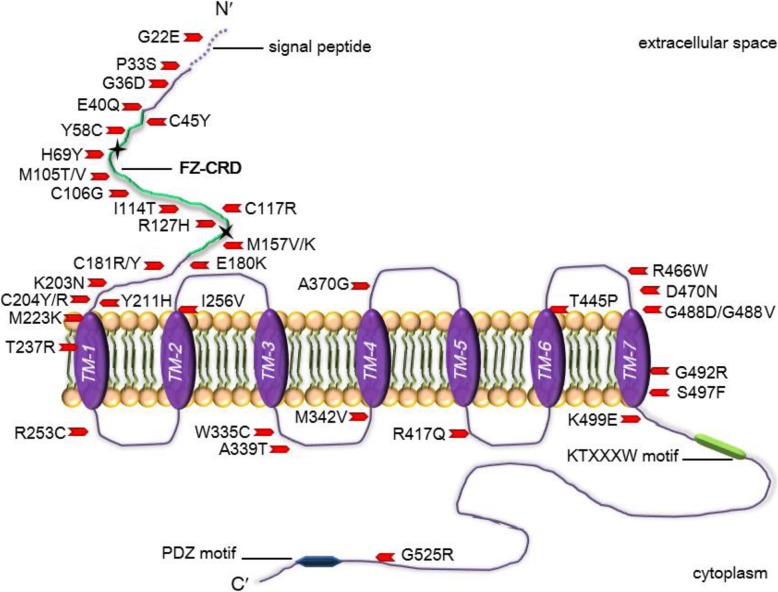
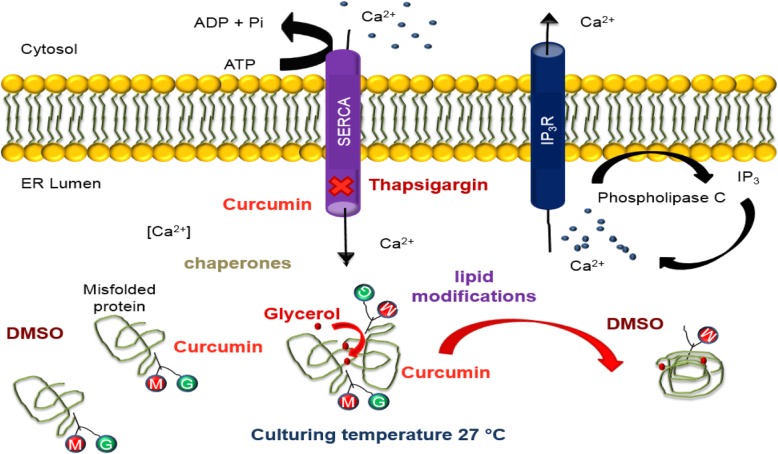
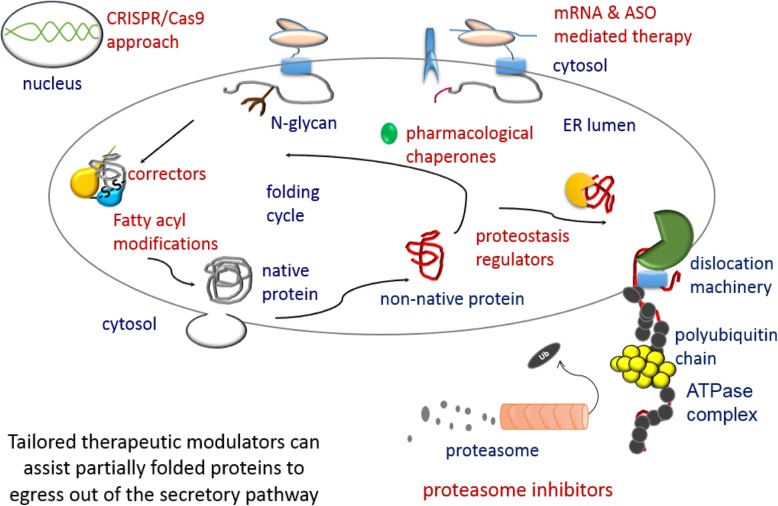
The ER is hub for protein folding. Proteins that harbor a Frizzled cysteine-rich domain (FZ-CRD) possess 10 conserved cysteine motifs held by a unique disulfide bridge pattern which attains a correct fold in the ER. Little is known about implications of disease-causing missense mutations within FZ-CRD families. Mutations in FZ-CRD of Frizzled class receptor 4 (FZD4) and Muscle, skeletal, receptor tyrosine kinase (MuSK) and Receptor tyrosine kinase-like orphan receptor 2 (ROR2) cause Familial Exudative Vitreoretinopathy (FEVR), Congenital Myasthenic Syndrome (CMS), and Robinow Syndrome (RS) respectively. We highlight reported pathogenic inherited missense mutations in FZ-CRD of FZD4, MuSK and ROR2 which misfold, and traffic abnormally in the ER, with ER-associated degradation (ERAD) as a common pathogenic mechanism for disease. Our review shows that all studied FZ-CRD mutants of RS, FEVR and CMS result in misfolded proteins and/or partially misfolded proteins with an ERAD fate, thus we coin them as "disorders of FZ-CRD". Abnormal trafficking was demonstrated in 17 of 29 mutants studied; 16 mutants were within and/or surrounding the FZ-CRD with two mutants distant from FZ-CRD. These ER-retained mutants were improperly N-glycosylated confirming ER-localization. FZD4 and MuSK mutants were tagged with polyubiquitin chains confirming targeting for proteasomal degradation. Investigating the cellular and molecular mechanisms of these mutations is important since misfolded protein and ER-targeted therapies are in development. The P344R-MuSK kinase mutant showed around 50% of its in-vitro autophosphorylation activity and P344R-MuSK increased two-fold on proteasome inhibition. M105T-FZD4, C204Y-FZD4, and P344R-MuSK mutants are thermosensitive and therefore, might benefit from extending the investigation to a larger number of chemical chaperones and/or proteasome inhibitors. Nonetheless, FZ-CRD ER-lipidation it less characterized in the literature and recent structural data sheds light on the importance of lipidation in protein glycosylation, proper folding, and ER trafficking. Current treatment strategies in-place for the conformational disease landscape is highlighted. From this review, we envision that disorders of FZ-CRD might be receptive to therapies that target FZ-CRD misfolding, regulation of fatty acids, and/or ER therapies; thus paving the way for a newly explored paradigm to treat different diseases with common defects.
Keywords: Cystic fibrosis conductance regulator protein; ERAD; protein misfolding; Familial exudative vitreoretinopathy; congenital myasthenic syndrome; Robinow syndrome; receptor tyrosine kinase-like orphan receptor 2; frizzled class receptor 4; muscle; Frizzled cysteine-rich domain; Frizzled receptors; Lipidation; Proteostasis; Receptor tyrosine kinase; conformational diseases; Skeletal; cis-unsaturated fatty acids.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Improved plasma membrane expression of the trafficking defective P344R mutant of muscle, skeletal, receptor tyrosine kinase (MuSK) causing congenital myasthenic syndrome.Int J Biochem Cell Biol. 2015 Mar;60:119-29. doi: 10.1016/j.biocel.2014.12.015. Epub 2015 Jan 3. Int J Biochem Cell Biol. 2015. PMID: 25562515
-
Identification of the cellular mechanisms that modulate trafficking of frizzled family receptor 4 (FZD4) missense mutants associated with familial exudative vitreoretinopathy.Invest Ophthalmol Vis Sci. 2014 Apr 17;55(6):3423-31. doi: 10.1167/iovs.14-13885. Invest Ophthalmol Vis Sci. 2014. PMID: 24744206
-
The evolutionary analysis reveals domain fusion of proteins with Frizzled-like CRD domain.Gene. 2014 Jan 1;533(1):229-39. doi: 10.1016/j.gene.2013.09.083. Epub 2013 Oct 14. Gene. 2014. PMID: 24135643
-
Endocrinopathies in the family of endoplasmic reticulum (ER) storage diseases: disorders of protein trafficking and the role of ER molecular chaperones.Endocr Rev. 1998 Apr;19(2):173-202. doi: 10.1210/edrv.19.2.0327. Endocr Rev. 1998. PMID: 9570036 Review.
-
Redox-dependent protein quality control in the endoplasmic reticulum: folding to degradation.Antioxid Redox Signal. 2012 May 15;16(10):1119-28. doi: 10.1089/ars.2011.4495. Epub 2012 Feb 23. Antioxid Redox Signal. 2012. PMID: 22229892 Review.
Cited by
-
Frizzled receptors: gatekeepers of Wnt signaling in development and disease.Front Cell Dev Biol. 2025 May 1;13:1599355. doi: 10.3389/fcell.2025.1599355. eCollection 2025. Front Cell Dev Biol. 2025. PMID: 40376615 Free PMC article. Review.
-
Role of NDP- and FZD4-Related Novel Mutations Identified in Patients with FEVR in Norrin/β-Catenin Signaling Pathway.Biomed Res Int. 2020 Apr 27;2020:7681926. doi: 10.1155/2020/7681926. eCollection 2020. Biomed Res Int. 2020. PMID: 32420371 Free PMC article.
-
Endoplasmic Reticulum Associated Protein Degradation (ERAD) in the Pathology of Diseases Related to TGFβ Signaling Pathway: Future Therapeutic Perspectives.Front Mol Biosci. 2020 Oct 29;7:575608. doi: 10.3389/fmolb.2020.575608. eCollection 2020. Front Mol Biosci. 2020. PMID: 33195419 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
