

Time to dissect the autoimmune etiology of cancer antibody immunotherapy
- PMID: 31895048
- PMCID: PMC6934191
- DOI: 10.1172/JCI131194
Time to dissect the autoimmune etiology of cancer antibody immunotherapy
Abstract
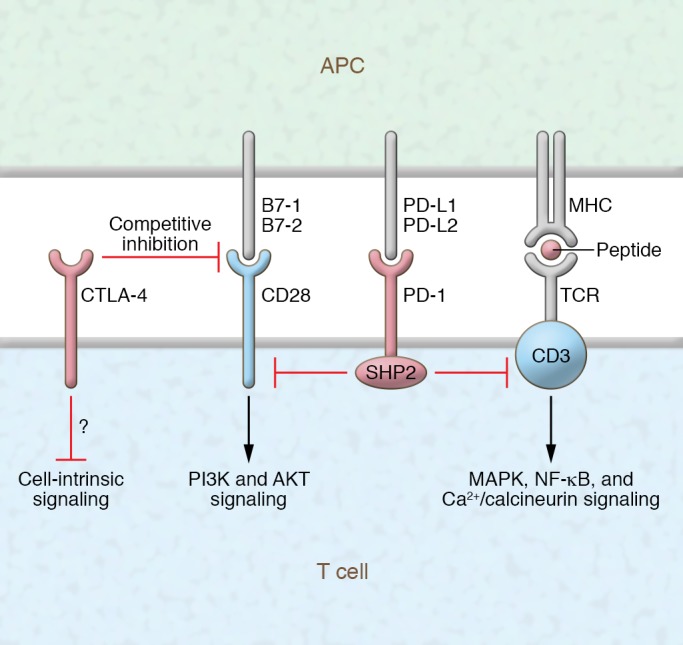
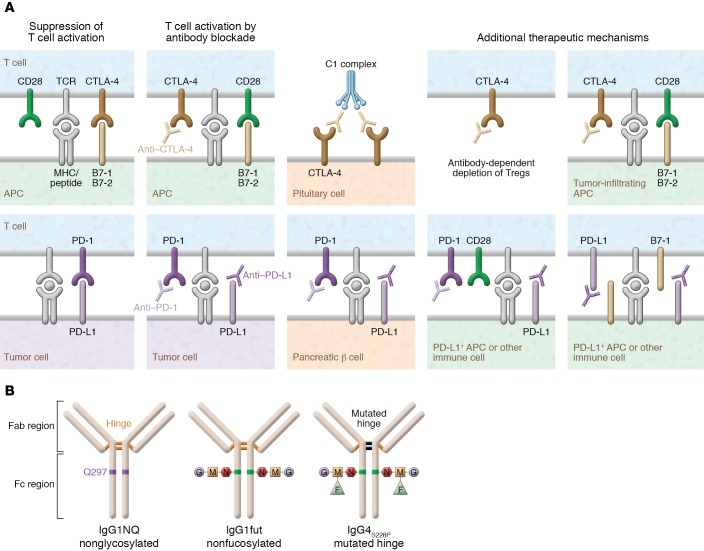
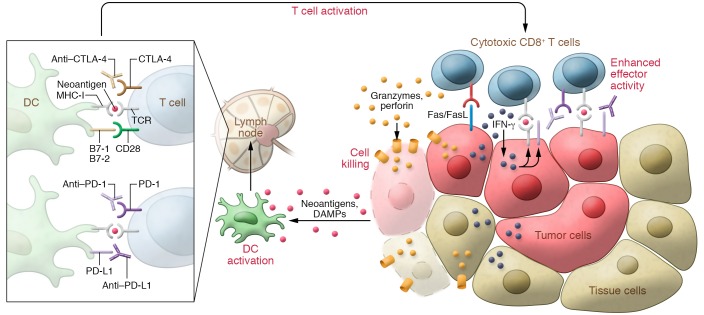
Immunotherapy has transformed the treatment landscape for a wide range of human cancers. Immune checkpoint inhibitors (ICIs), monoclonal antibodies that block the immune-regulatory "checkpoint" receptors CTLA-4, PD-1, or its ligand PD-L1, can produce durable responses in some patients. However, coupled with their success, these treatments commonly evoke a wide range of immune-related adverse events (irAEs) that can affect any organ system and can be treatment-limiting and life-threatening, such as diabetic ketoacidosis, which appears to be more frequent than initially described. The majority of irAEs from checkpoint blockade involve either barrier tissues (e.g., gastrointestinal mucosa or skin) or endocrine organs, although any organ system can be affected. Often, irAEs resemble spontaneous autoimmune diseases, such as inflammatory bowel disease, autoimmune thyroid disease, type 1 diabetes mellitus (T1D), and autoimmune pancreatitis. Yet whether similar molecular or pathologic mechanisms underlie these apparent autoimmune adverse events and classical autoimmune diseases is presently unknown. Interestingly, evidence links HLA alleles associated with high risk for autoimmune disease with ICI-induced T1D and colitis. Understanding the genetic risks and immunologic mechanisms driving ICI-mediated inflammatory toxicities may not only identify therapeutic targets useful for managing irAEs, but may also provide new insights into the pathoetiology and treatment of autoimmune diseases.
Conflict of interest statement
Figures
References
-
- Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8(9):1069–1086. doi: 10.1158/2159-8290.CD-18-0367. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
