

Genome-wide analysis identifies cis-acting elements regulating mRNA polyadenylation and translation during vertebrate oocyte maturation
- PMID: 31896558
- PMCID: PMC7025505
- DOI: 10.1261/rna.073247.119
Genome-wide analysis identifies cis-acting elements regulating mRNA polyadenylation and translation during vertebrate oocyte maturation
Abstract
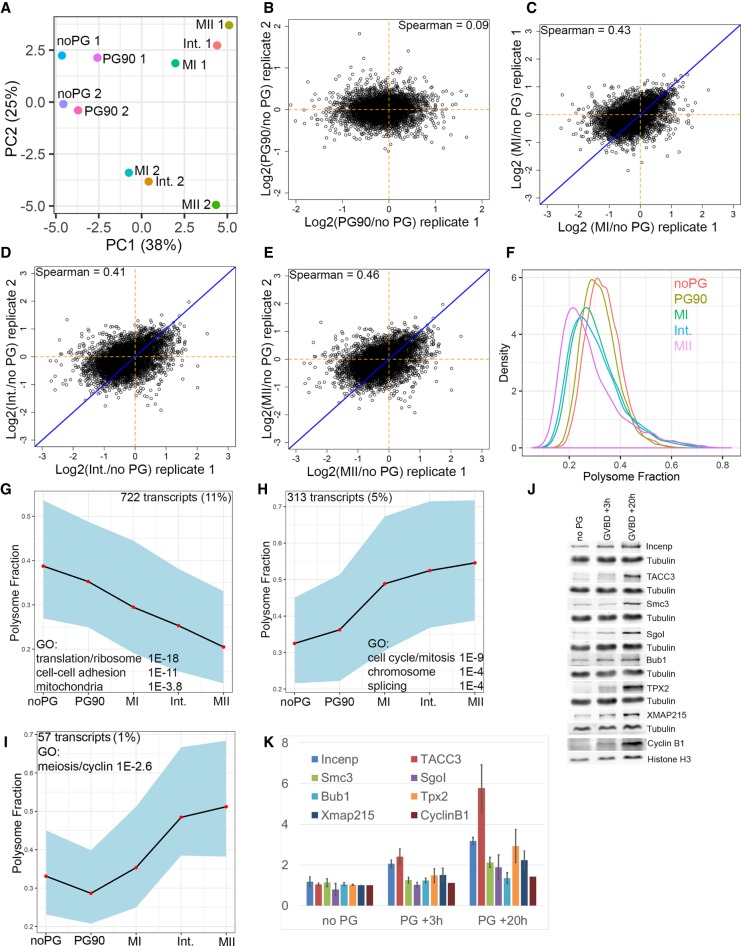
Most cells change patterns of gene expression through transcriptional regulation. In contrast, oocytes are transcriptionally silent and regulate mRNA poly(A) tail length to control protein production. However, the genome-wide relationship of poly(A) tail changes to mRNA translation during vertebrate oocyte maturation is not known. We used Tail-seq and polyribosome analysis to measure poly(A) tail and translational changes during oocyte maturation in Xenopus laevis We identified large-scale poly(A) and translational changes during oocyte maturation, with poly(A) tail length changes preceding translational changes. Proteins important for completion of the meiotic divisions and early development exhibited increased polyadenylation and translation during oocyte maturation. A family of U-rich sequence elements was enriched near the polyadenylation signal of polyadenylated and translationally activated mRNAs. We propose that changes in mRNA polyadenylation are a conserved mechanism regulating protein expression during vertebrate oocyte maturation and that these changes are controlled by a spatial code of cis-acting sequence elements.
Keywords: oocyte; polyadenylation; translation.
© 2020 Yang et al.; Published by Cold Spring Harbor Laboratory Press for the RNA Society.
Figures







References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases