

Improved protein structure prediction using predicted interresidue orientations
- PMID: 31896580
- PMCID: PMC6983395
- DOI: 10.1073/pnas.1914677117
Improved protein structure prediction using predicted interresidue orientations
Abstract
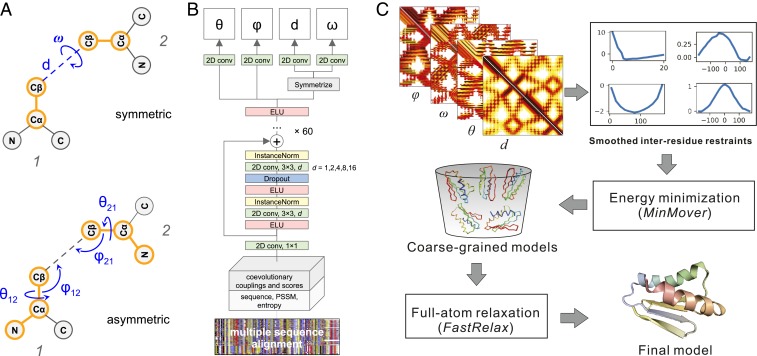
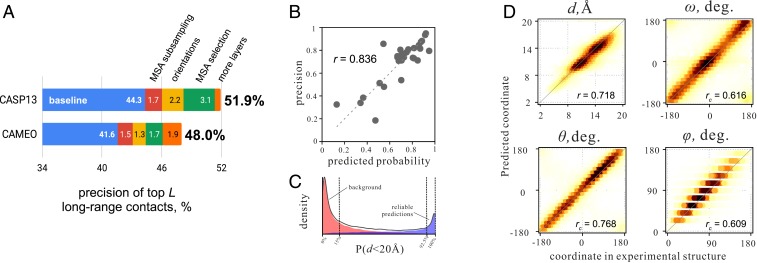
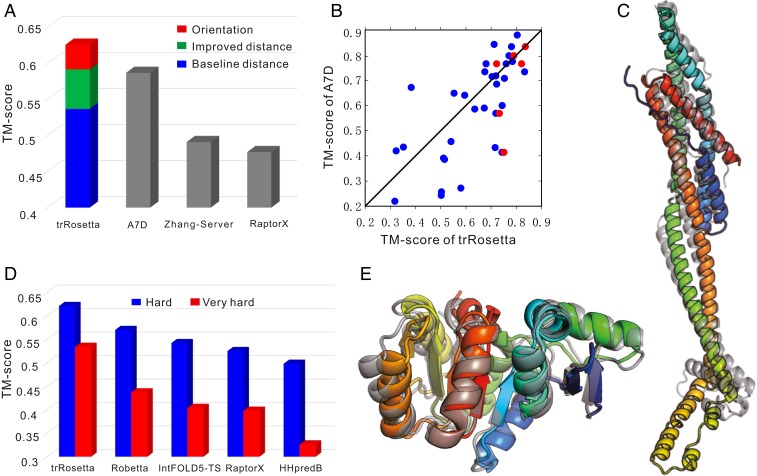
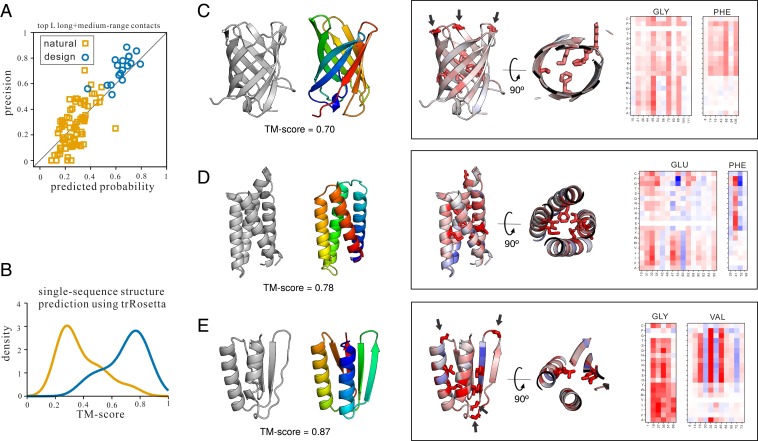
The prediction of interresidue contacts and distances from coevolutionary data using deep learning has considerably advanced protein structure prediction. Here, we build on these advances by developing a deep residual network for predicting interresidue orientations, in addition to distances, and a Rosetta-constrained energy-minimization protocol for rapidly and accurately generating structure models guided by these restraints. In benchmark tests on 13th Community-Wide Experiment on the Critical Assessment of Techniques for Protein Structure Prediction (CASP13)- and Continuous Automated Model Evaluation (CAMEO)-derived sets, the method outperforms all previously described structure-prediction methods. Although trained entirely on native proteins, the network consistently assigns higher probability to de novo-designed proteins, identifying the key fold-determining residues and providing an independent quantitative measure of the "ideality" of a protein structure. The method promises to be useful for a broad range of protein structure prediction and design problems.
Keywords: deep learning; protein contact prediction; protein structure prediction.
Conflict of interest statement
The authors declare no competing interest.
Figures
Comment in
-
Deep learning 3D structures.Nat Methods. 2020 Mar;17(3):249. doi: 10.1038/s41592-020-0779-y. Nat Methods. 2020. PMID: 32132733 No abstract available.
References
-
- Abriata L. A., Tamò G. E., Dal Peraro M., A further leap of improvement in tertiary structure prediction in CASP13 prompts new routes for future assessments. Proteins 87, 1100–1112 (2019). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
