

Autophagy and microbial pathogenesis
- PMID: 31896796
- PMCID: PMC7205878
- DOI: 10.1038/s41418-019-0481-8
Autophagy and microbial pathogenesis
Abstract
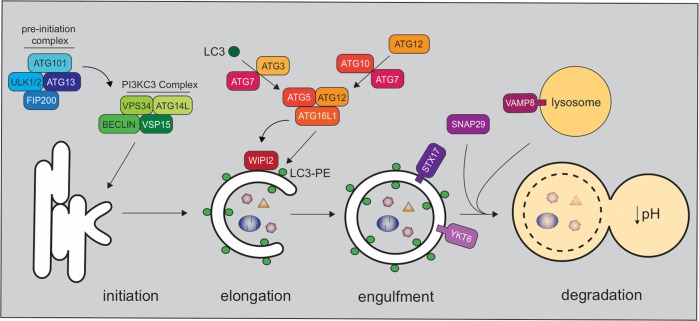
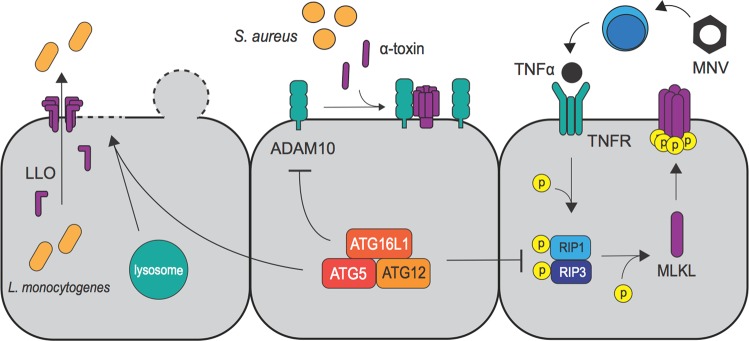
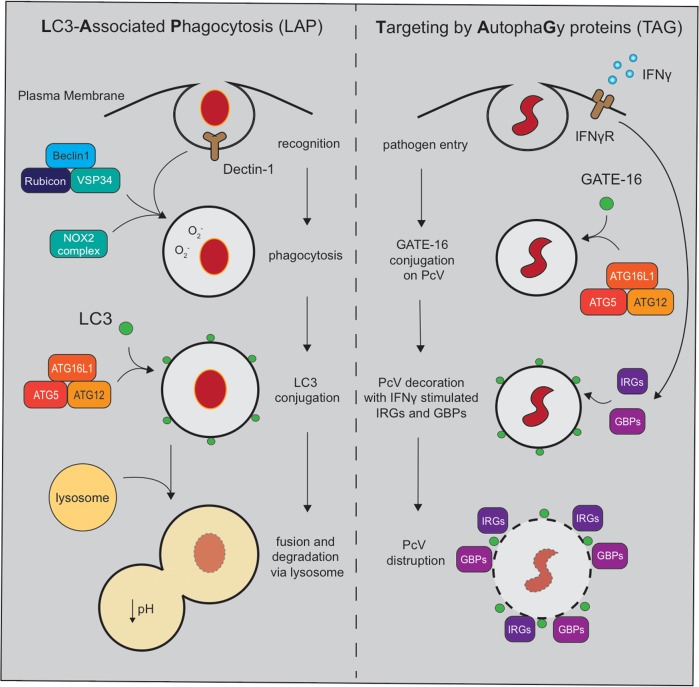
Autophagy is a cell biological process that promotes resilience in the face of environmental perturbations. Given that infectious agents represent a major type of environmental threat, it follows that the autophagy pathway is central to the outcome of host-microbe interactions. Detailed molecular studies have revealed intricate ways in which autophagy suppresses or enhances the fitness of infectious agents, particularly intracellular pathogens such as viruses that require the host cell machinery for replication. Findings in animal models have reinforced the importance of these events that occur within individual cells and have extended the role of autophagy to extracellular microbes and immunity at the whole organism level. These functions impact adaptation to bacteria that are part of the gut microbiota, which has implications for the etiology of chronic disorders such as inflammatory bowel disease. Despite major advances in how autophagy regulates inflammatory reactions toward microbes, many challenges remain, including distinguishing autophagy from closely related pathways such as LC3-associated phagocytosis. Here, we review the role of autophagy in microbial pathogenesis at the level of organismal biology. In addition to providing an overview of the prominent function of autophagy proteins in host-microbe interactions, we highlight how observations at the cellular level are informing pathogenesis studies and offer our perspective on the future directions of the field.
Conflict of interest statement
VJT is an inventor on patents and patent applications filed by New York University, which are currently under commercial license to Janssen Biotech Inc. KC has consulted for or received an honorarium from Puretech Health, Genentech, and AbbVie, Inc., has received research support from Puretech Health and Pfizer, Inc, and has a provisional patent, US Patent Appln. No. 15/625,934.
Figures
References
Publication types
MeSH terms
Grants and funding
- UL1 TR001445/TR/NCATS NIH HHS/United States
- P30 CA016087/CA/NCI NIH HHS/United States
- R01 DK093668/DK/NIDDK NIH HHS/United States
- P01 CA023766/CA/NCI NIH HHS/United States
- F31 HL137304/HL/NHLBI NIH HHS/United States
- R01 AI105129/AI/NIAID NIH HHS/United States
- R01 DK103788/DK/NIDDK NIH HHS/United States
- R01 HL123340/HL/NHLBI NIH HHS/United States
- R01 AI099394/AI/NIAID NIH HHS/United States
- R01 HL125816/HL/NHLBI NIH HHS/United States
- R01 AI130945/AI/NIAID NIH HHS/United States
- T32 AI007180/AI/NIAID NIH HHS/United States
- R01 AI121244/AI/NIAID NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
LinkOut - more resources
Full Text Sources
