

Evolutionary superscaffolding and chromosome anchoring to improve Anopheles genome assemblies
- PMID: 31898513
- PMCID: PMC6939337
- DOI: 10.1186/s12915-019-0728-3
Evolutionary superscaffolding and chromosome anchoring to improve Anopheles genome assemblies
Abstract
Background: New sequencing technologies have lowered financial barriers to whole genome sequencing, but resulting assemblies are often fragmented and far from 'finished'. Updating multi-scaffold drafts to chromosome-level status can be achieved through experimental mapping or re-sequencing efforts. Avoiding the costs associated with such approaches, comparative genomic analysis of gene order conservation (synteny) to predict scaffold neighbours (adjacencies) offers a potentially useful complementary method for improving draft assemblies.
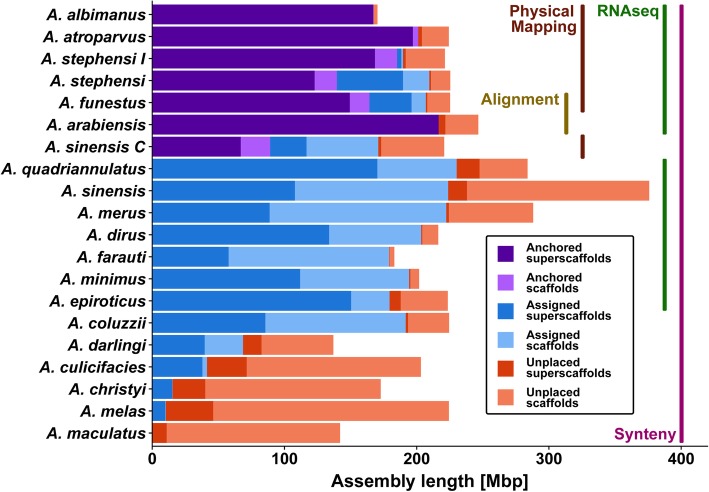
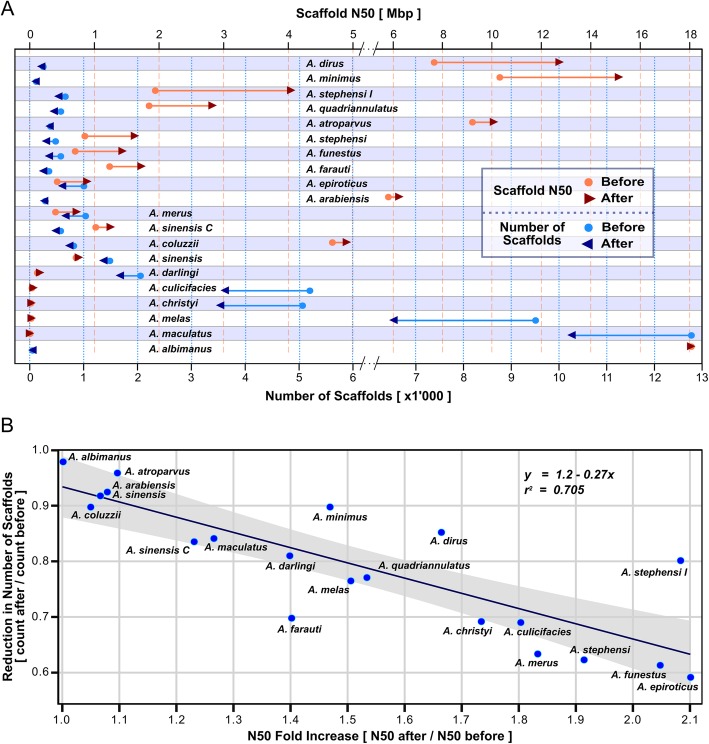
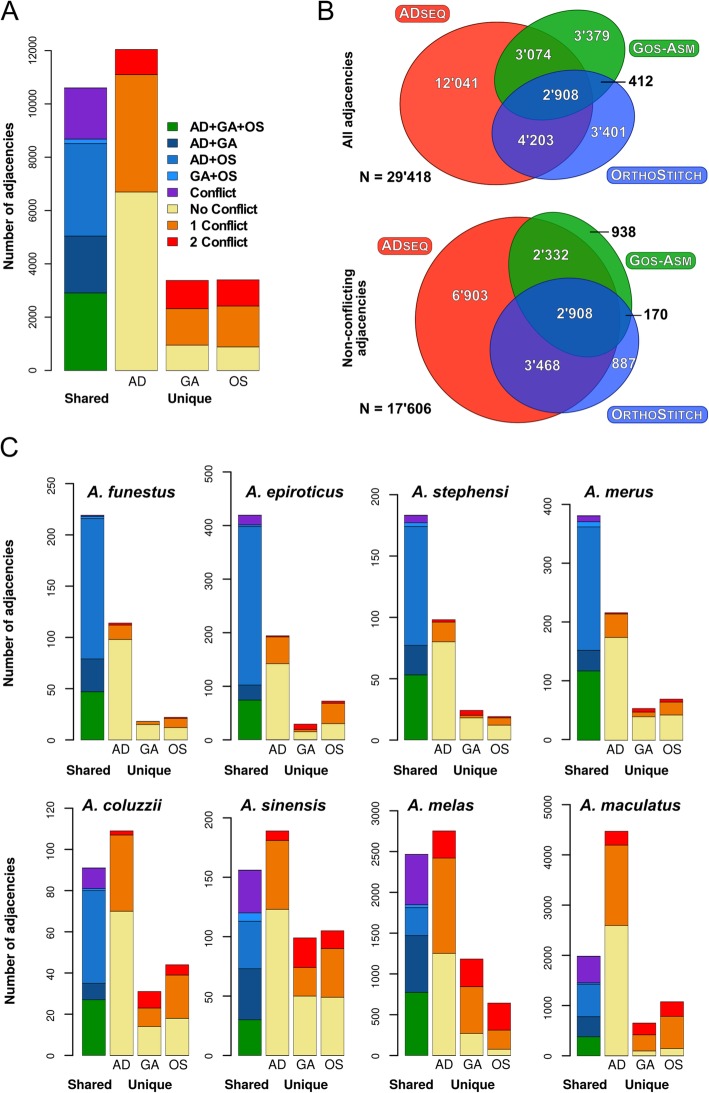
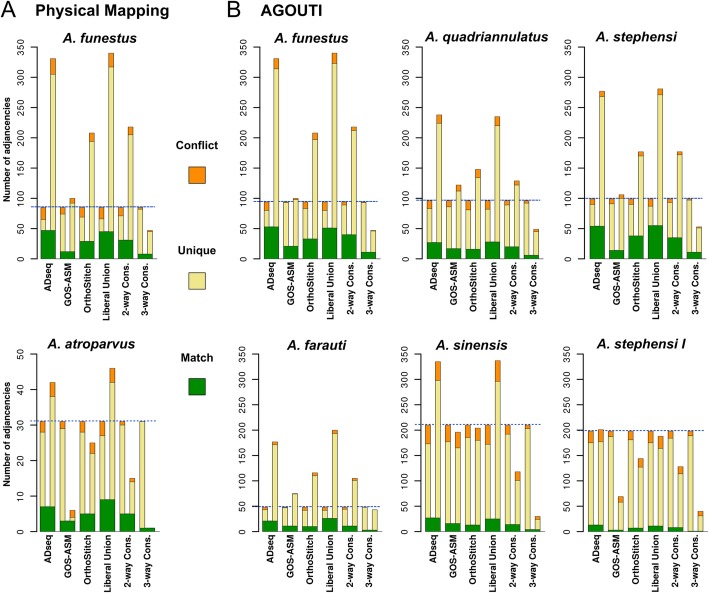
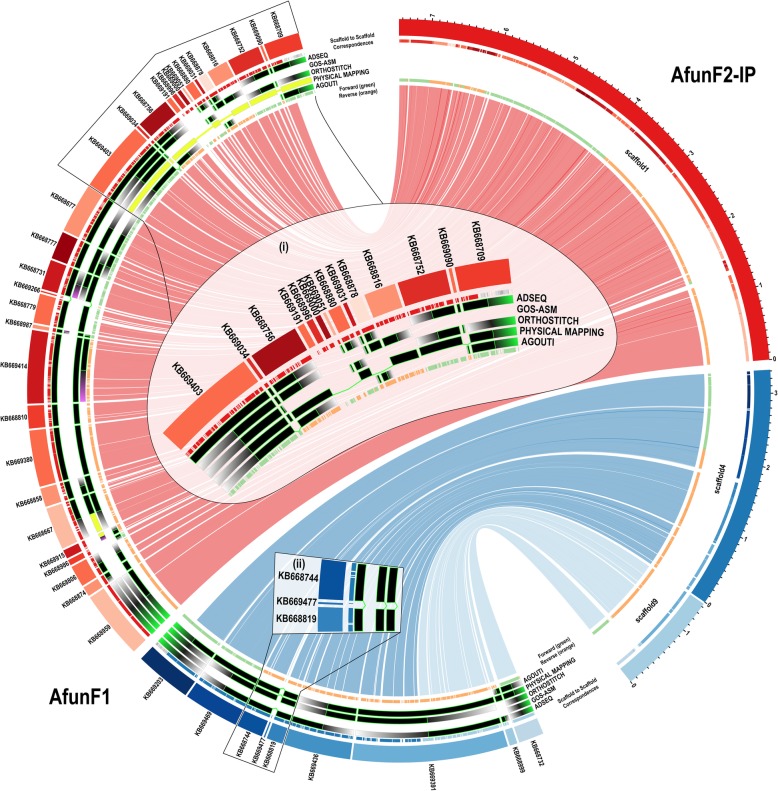
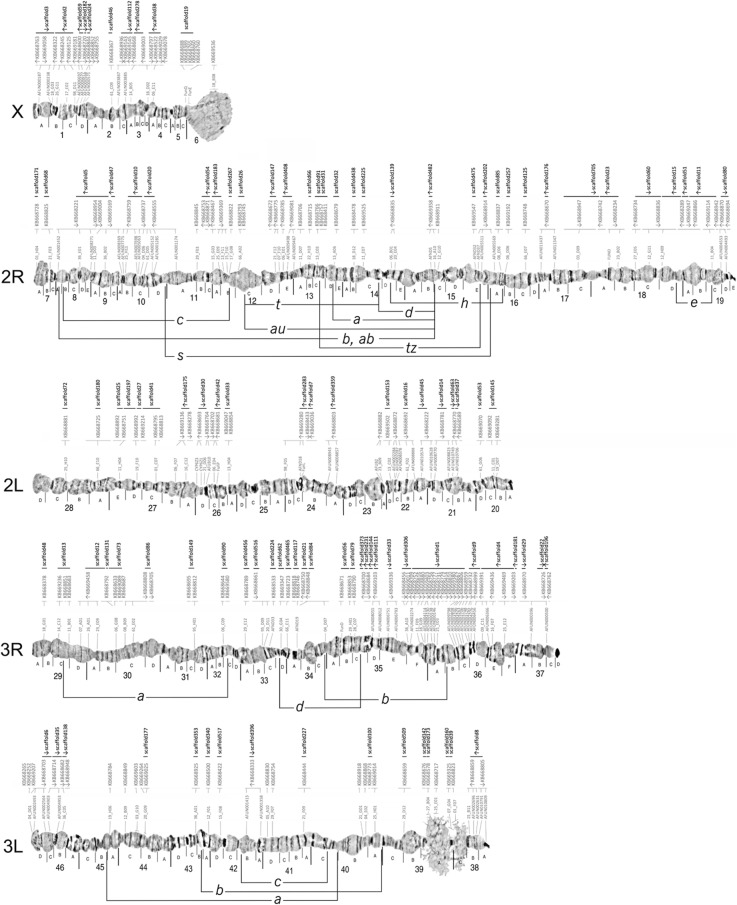
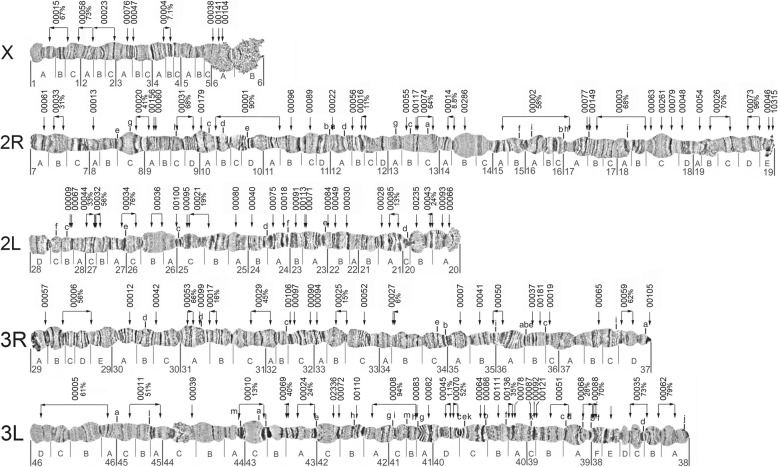
Results: We evaluated and employed 3 gene synteny-based methods applied to 21 Anopheles mosquito assemblies to produce consensus sets of scaffold adjacencies. For subsets of the assemblies, we integrated these with additional supporting data to confirm and complement the synteny-based adjacencies: 6 with physical mapping data that anchor scaffolds to chromosome locations, 13 with paired-end RNA sequencing (RNAseq) data, and 3 with new assemblies based on re-scaffolding or long-read data. Our combined analyses produced 20 new superscaffolded assemblies with improved contiguities: 7 for which assignments of non-anchored scaffolds to chromosome arms span more than 75% of the assemblies, and a further 7 with chromosome anchoring including an 88% anchored Anopheles arabiensis assembly and, respectively, 73% and 84% anchored assemblies with comprehensively updated cytogenetic photomaps for Anopheles funestus and Anopheles stephensi.
Conclusions: Experimental data from probe mapping, RNAseq, or long-read technologies, where available, all contribute to successful upgrading of draft assemblies. Our evaluations show that gene synteny-based computational methods represent a valuable alternative or complementary approach. Our improved Anopheles reference assemblies highlight the utility of applying comparative genomics approaches to improve community genomic resources.
Keywords: Bioinformatics; Chromosomes; Comparative genomics; Computational evolutionary biology; Gene synteny; Genome assembly; Mosquito genomes; Orthology; Physical mapping.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
