

Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia
- PMID: 31899794
- PMCID: PMC7046598
- DOI: 10.1182/blood.2019003140
Antibodies against the erythroferrone N-terminal domain prevent hepcidin suppression and ameliorate murine thalassemia
Abstract
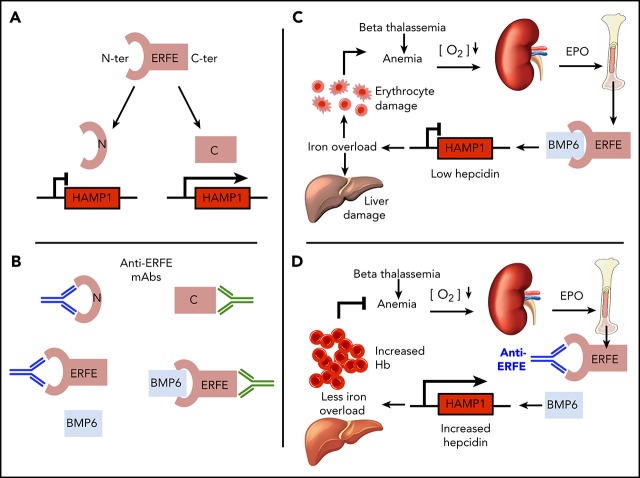
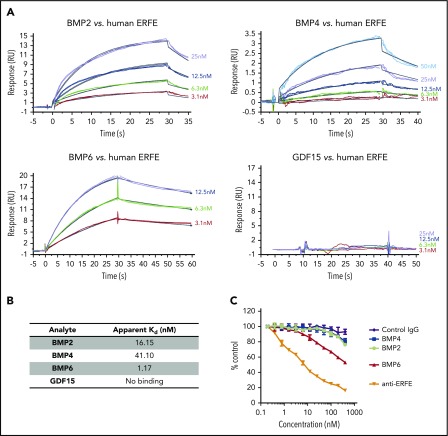
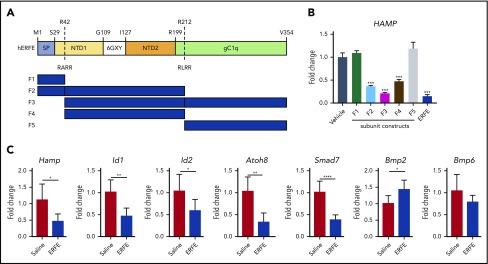
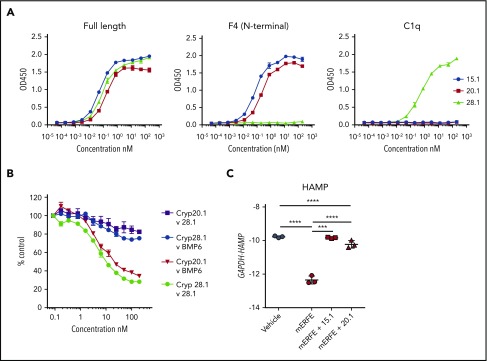
Erythroferrone (ERFE) is produced by erythroblasts in response to erythropoietin (EPO) and acts in the liver to prevent hepcidin stimulation by BMP6. Hepcidin suppression allows for the mobilization of iron to the bone marrow for the production of red blood cells. Aberrantly high circulating ERFE in conditions of stress erythropoiesis, such as in patients with β-thalassemia, promotes the tissue iron accumulation that substantially contributes to morbidity in these patients. Here we developed antibodies against ERFE to prevent hepcidin suppression and to correct the iron loading phenotype in a mouse model of β-thalassemia [Hbb(th3/+) mice] and used these antibodies as tools to further characterize ERFE's mechanism of action. We show that ERFE binds to BMP6 with nanomolar affinity and binds BMP2 and BMP4 with somewhat weaker affinities. We found that BMP6 binds the N-terminal domain of ERFE, and a polypeptide derived from the N terminus of ERFE was sufficient to cause hepcidin suppression in Huh7 hepatoma cells and in wild-type mice. Anti-ERFE antibodies targeting the N-terminal domain prevented hepcidin suppression in ERFE-treated Huh7 cells and in EPO-treated mice. Finally, we observed a decrease in splenomegaly and serum and liver iron in anti-ERFE-treated Hbb(th3/+) mice, accompanied by an increase in red blood cells and hemoglobin and a decrease in reticulocyte counts. In summary, we show that ERFE binds BMP6 directly and with high affinity, and that antibodies targeting the N-terminal domain of ERFE that prevent ERFE-BMP6 interactions constitute a potential therapeutic tool for iron loading anemias.
© 2020 by The American Society of Hematology.
Conflict of interest statement
Conflict-of-interest disclosure: This work was supported in part by funding from Pfizer to J.A., K.M., D.Q., S.J.D., and H.D. N.F., A.S., S.B., E.L., M.S.T., D.D.P., O.C., M.L., J.E.M., and R.J. are employed by Pfizer. N.F., O.C., R.J., J.A., K.M., S.J.D., and H.D. are named inventors on a patent application currently under evaluation. The remaining authors declare no competing financial interests.
Figures

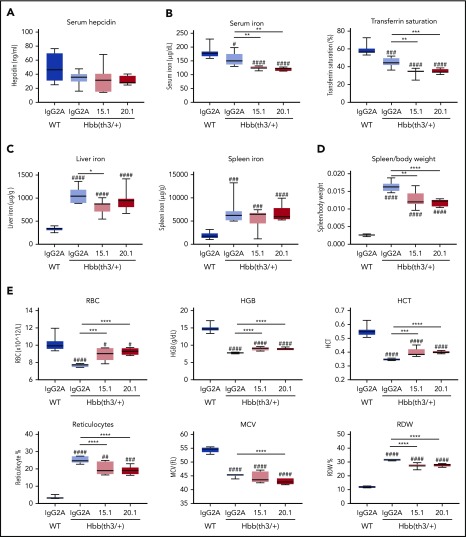
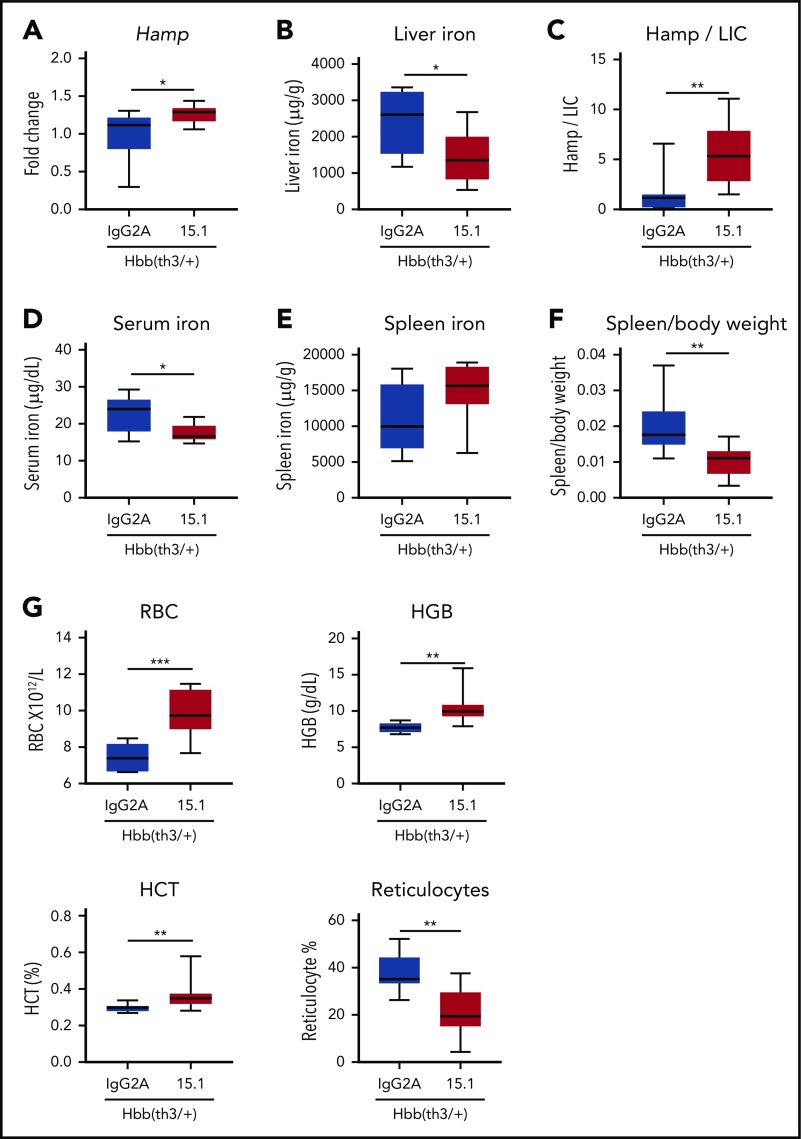
Comment in
-
Drugging erythroferrone to treat anemias.Blood. 2020 Feb 20;135(8):516-518. doi: 10.1182/blood.2019004678. Blood. 2020. PMID: 32078688 No abstract available.
References
-
- Rachmilewitz EA, Giardina PJ. How I treat thalassemia. Blood. 2011;118(13):3479-3488. - PubMed
-
- Taher AT, Weatherall DJ, Cappellini MD. Thalassaemia. Lancet. 2018;391(10116):155-167. - PubMed
-
- Flint J, Harding RM, Boyce AJ, Clegg JB. The population genetics of the haemoglobinopathies. Baillieres Clin Haematol. 1998;11(1):1-51. - PubMed
-
- Tantiworawit A, Charoenkwan P, Hantrakool S, Choeyprasert W, Sivasomboon C, Sanguansermsri T. Iron overload in non-transfusion-dependent thalassemia: association with genotype and clinical risk factors. Int J Hematol. 2016;103(6):643-648. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
