

Insights into the prenatal origin of childhood acute lymphoblastic leukemia
- PMID: 31902036
- PMCID: PMC7098935
- DOI: 10.1007/s10555-019-09841-1
Insights into the prenatal origin of childhood acute lymphoblastic leukemia
Abstract
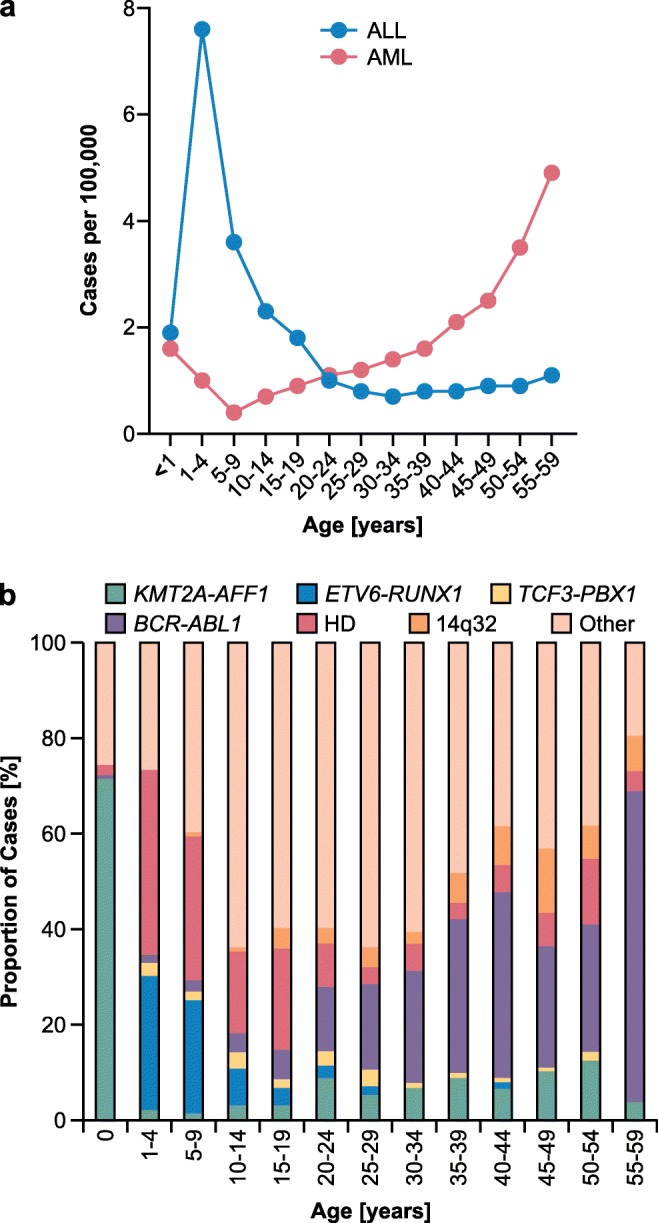
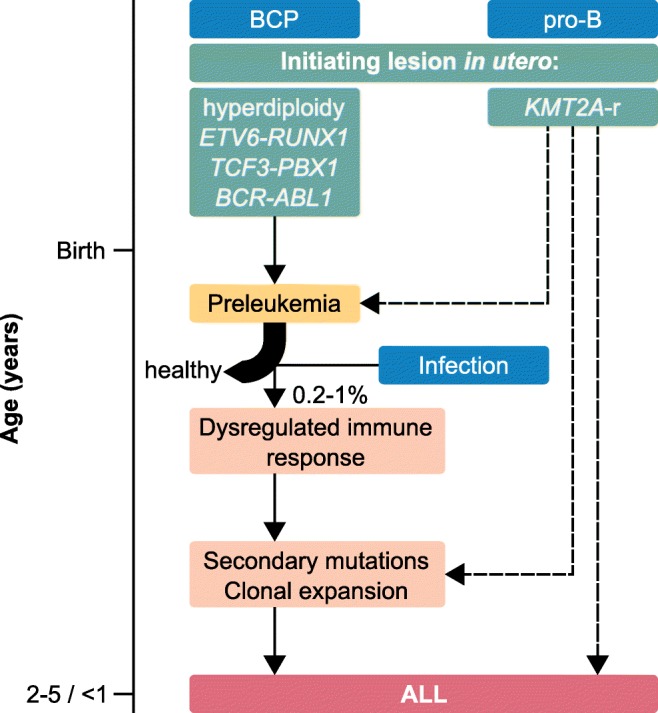
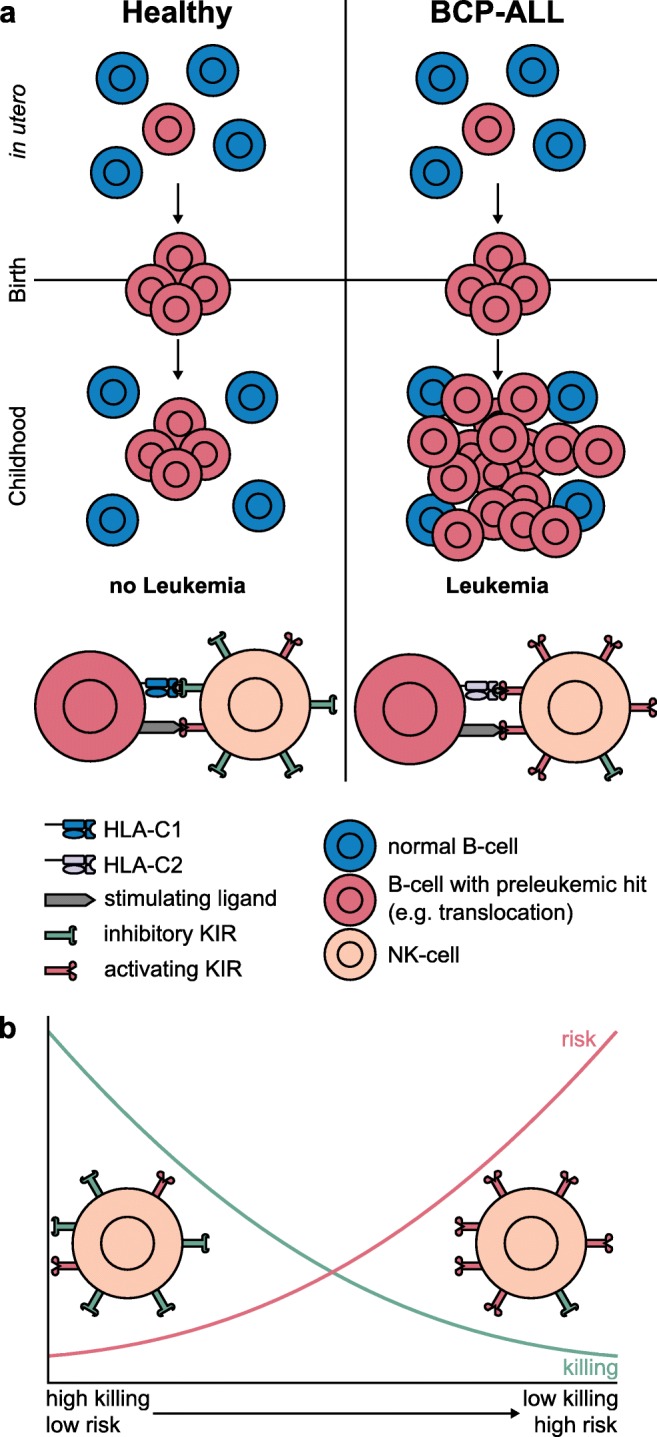
Pediatric acute lymphoblastic leukemia (ALL) is defined by recurrent chromosomal aberrations including hyperdiploidy and chromosomal translocations. Many of these aberrations originate in utero and the cells transform in early childhood through acquired secondary mutations. In this review, we will discuss the most common prenatal lesions that can lead to childhood ALL, with a special emphasis on the most common translocation in childhood ALL, t(12;21), which results in the ETV6-RUNX1 gene fusion. The ETV6-RUNX1 fusion arises prenatally and at a 500-fold higher frequency than the corresponding ALL. Even though the findings regarding the frequency of ETV6-RUNX1 were originally challenged, newer studies have confirmed the higher frequency. The prenatal origin has also been proven for other gene fusions, including KMT2A, the translocations t(1;19) and t(9;22) leading to TCF3-PBX1 and BCR-ABL1, respectively, as well as high hyperdiploidy. For most of these aberrations, there is evidence for more frequent occurrence than the corresponding leukemia incidences. We will briefly discuss what is known about the cells of origin, the mechanisms of leukemic transformation through lack of immunosurveillance, and why only a part of the carriers develops ALL.
Keywords: ALL; Fusion genes; Preleukemia; Prenatal origin.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
High incidence of RAS pathway mutations among sentinel genetic lesions of Korean pediatric BCR-ABL1-like acute lymphoblastic leukemia.Cancer Med. 2020 Jul;9(13):4632-4639. doi: 10.1002/cam4.3099. Epub 2020 May 7. Cancer Med. 2020. PMID: 32378810 Free PMC article.
-
Flow cytometric predictive scoring systems for common fusions ETV6/RUNX1, BCR/ABL1, TCF3/PBX1 and rearrangements of the KMT2A gene, proposed for the initial cytogenetic approach in cases of B-acute lymphoblastic leukemia.Int J Lab Hematol. 2019 Jun;41(3):364-372. doi: 10.1111/ijlh.12983. Epub 2019 Feb 7. Int J Lab Hematol. 2019. PMID: 30730614
-
Two novel fusion genes, AIF1L-ETV6 and ABL1-AIF1L, result together with ETV6-ABL1 from a single chromosomal rearrangement in acute lymphoblastic leukemia with prenatal origin.Genes Chromosomes Cancer. 2018 Sep;57(9):471-477. doi: 10.1002/gcc.6. Epub 2018 Jul 30. Genes Chromosomes Cancer. 2018. PMID: 29726059
-
Targeting signaling pathways in acute lymphoblastic leukemia: new insights.Hematology Am Soc Hematol Educ Program. 2013;2013:118-25. doi: 10.1182/asheducation-2013.1.118. Hematology Am Soc Hematol Educ Program. 2013. PMID: 24319172 Review.
-
The Landscape of Secondary Genetic Rearrangements in Pediatric Patients with B-Cell Acute Lymphoblastic Leukemia with t(12;21).Cells. 2023 Jan 18;12(3):357. doi: 10.3390/cells12030357. Cells. 2023. PMID: 36766699 Free PMC article. Review.
Cited by
-
Hyperdiploidy: the longest known, most prevalent, and most enigmatic form of acute lymphoblastic leukemia in children.Leukemia. 2022 Dec;36(12):2769-2783. doi: 10.1038/s41375-022-01720-z. Epub 2022 Oct 20. Leukemia. 2022. PMID: 36266323 Free PMC article. Review.
-
Prenatal Origin of Pediatric Leukemia: Lessons From Hematopoietic Development.Front Cell Dev Biol. 2021 Jan 12;8:618164. doi: 10.3389/fcell.2020.618164. eCollection 2020. Front Cell Dev Biol. 2021. PMID: 33511126 Free PMC article. Review.
-
T cell exhaustion in pediatric B-ALL: current knowledge and future perspectives.Front Immunol. 2025 May 28;16:1531145. doi: 10.3389/fimmu.2025.1531145. eCollection 2025. Front Immunol. 2025. PMID: 40503229 Free PMC article. Review.
-
Genome-wide CRISPR screens identify ferroptosis as a novel therapeutic vulnerability in acute lymphoblastic leukemia.Haematologica. 2023 Feb 1;108(2):382-393. doi: 10.3324/haematol.2022.280786. Haematologica. 2023. PMID: 36134452 Free PMC article.
-
Modulation of the ETV6::RUNX1 Gene Fusion Prevalence in Newborns by Corticosteroid Use During Pregnancy.Int J Mol Sci. 2025 Mar 25;26(7):2971. doi: 10.3390/ijms26072971. Int J Mol Sci. 2025. PMID: 40243620 Free PMC article. Clinical Trial.
References
-
- Howlader N, Noone AM, Krapcho M, Miller D, Brest A, Yu M, et al. SEER cancer statistics review, 1975–2016. Bethesda, MD: National Cancer Institute; 2019.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
