

A tumor suppressor enhancing module orchestrated by GATA4 denotes a therapeutic opportunity for GATA4 deficient HCC patients
- PMID: 31903133
- PMCID: PMC6929984
- DOI: 10.7150/thno.38060
A tumor suppressor enhancing module orchestrated by GATA4 denotes a therapeutic opportunity for GATA4 deficient HCC patients
Abstract
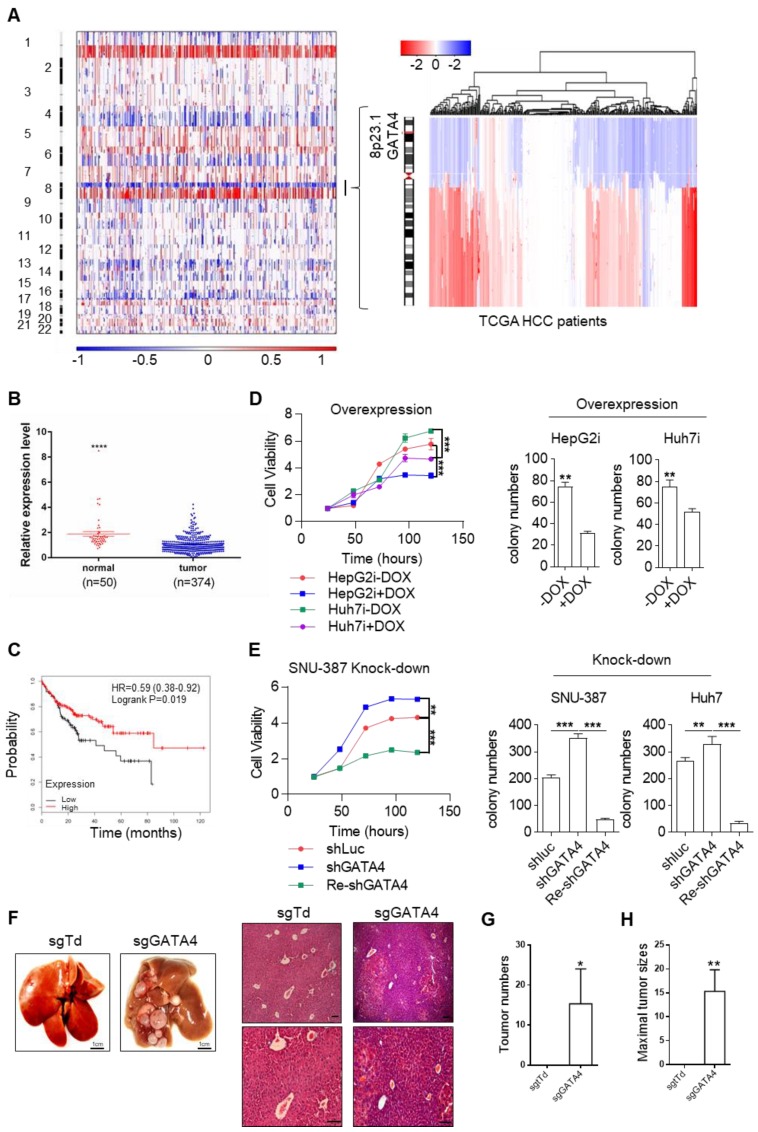
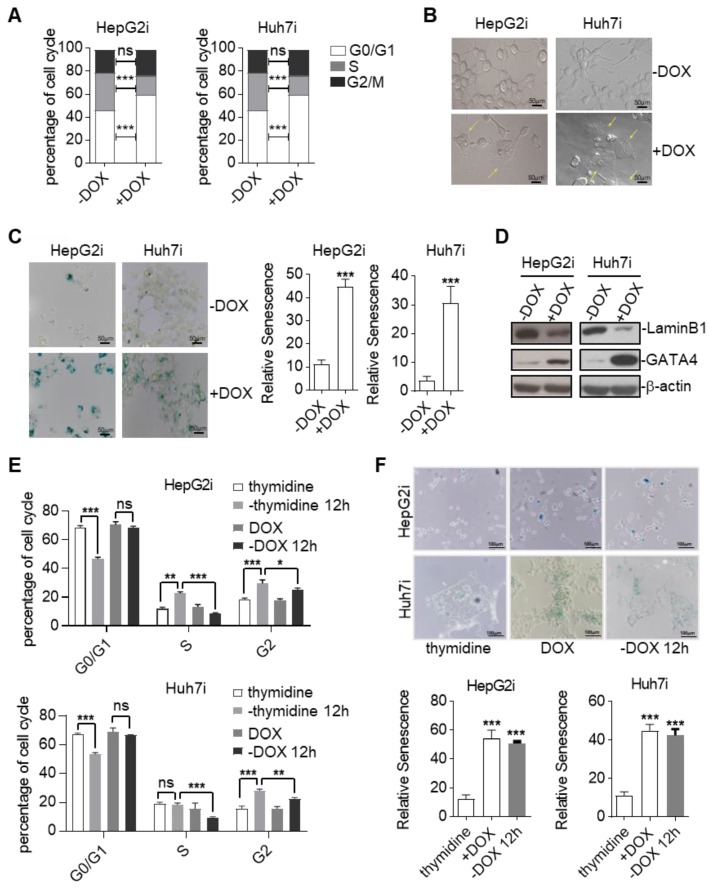
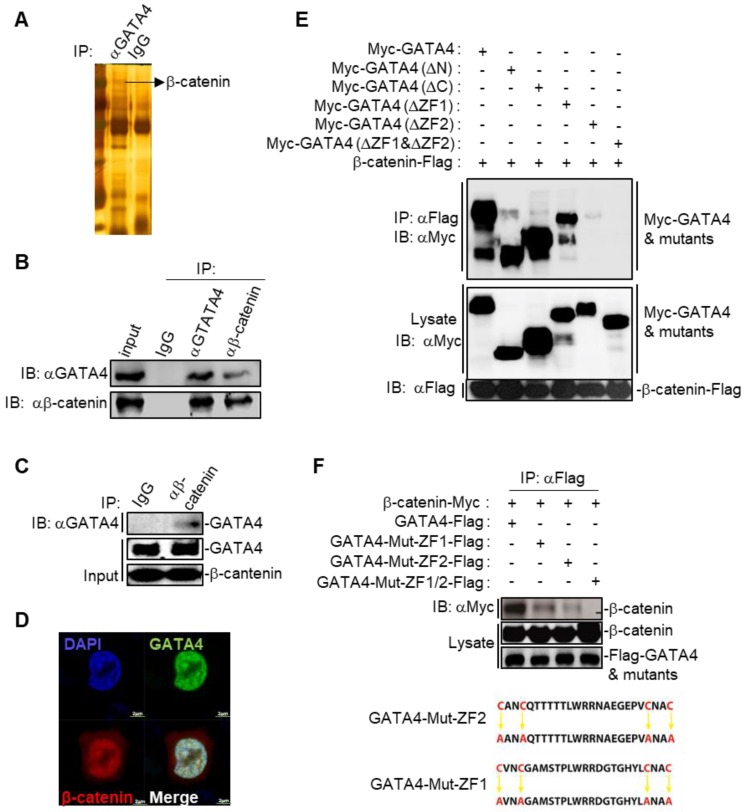
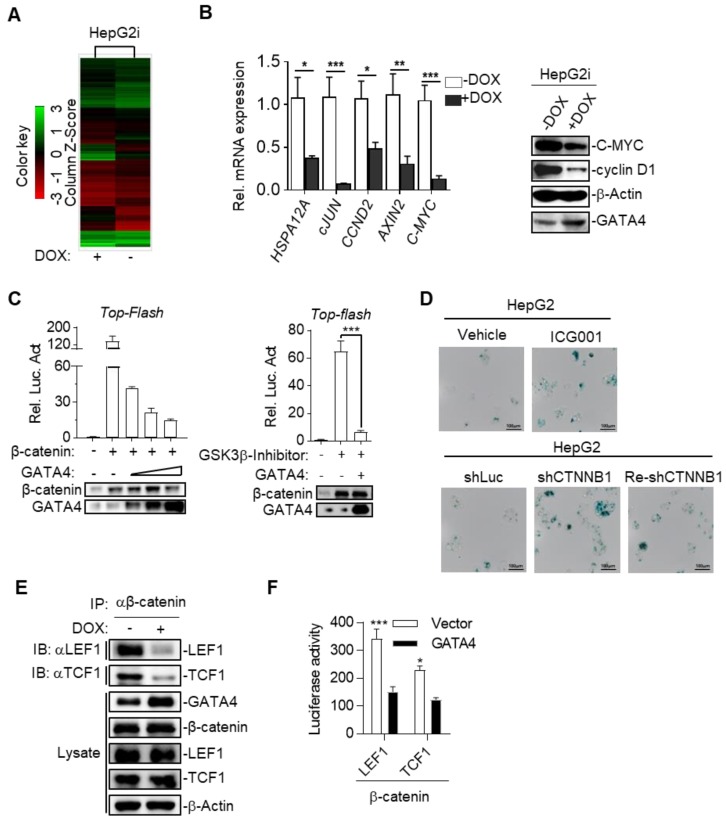
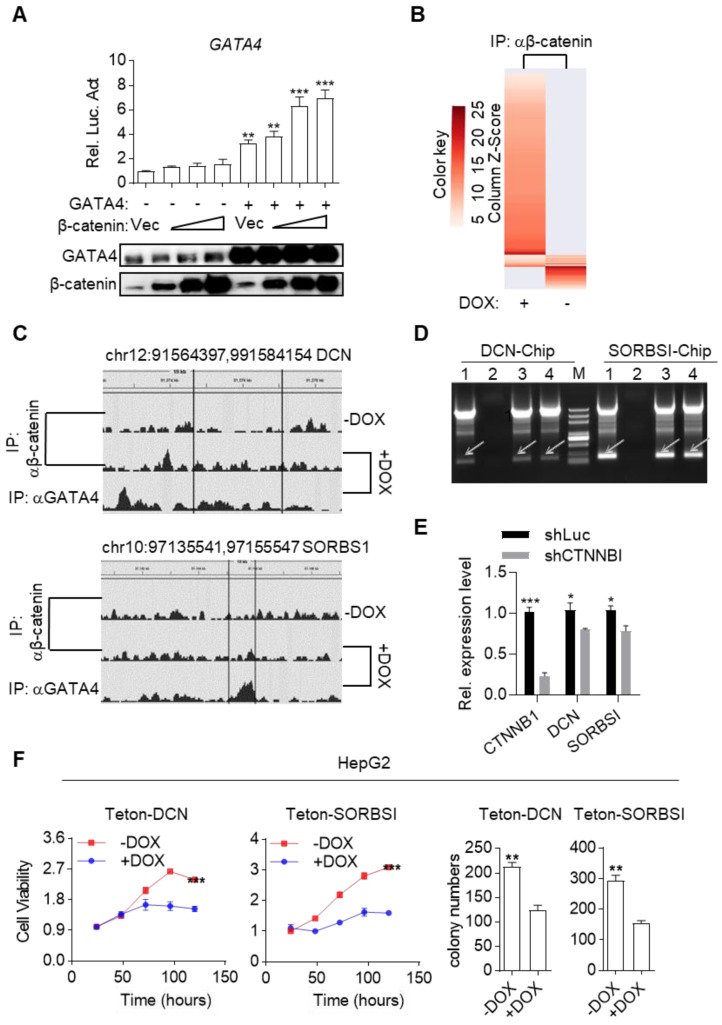
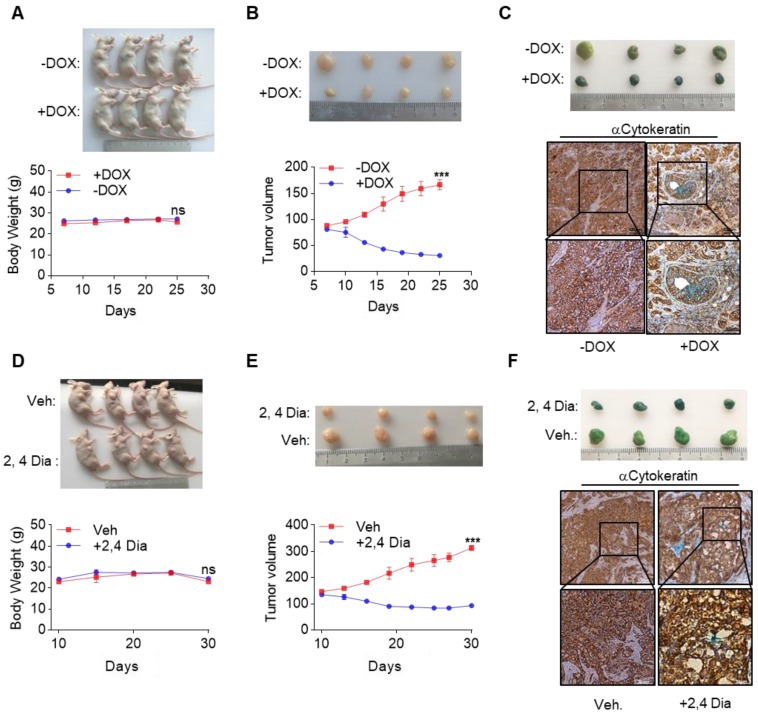
Rationale: Effective targeting therapies are limited in Hepatocellular carcinoma (HCC) clinic. Characterization of tumor suppressor genes (TSGs) and elucidation their signaling cascades could shed light on new strategies for developing targeting therapies for HCC. Methods: We checked genome-wide DNA copy number variation (CNV) of HCC samples, focusing on deleted genes for TSG candidates. Clinical data, in vitro and in vivo data were collected to validate the tumor suppressor functions. Results: Focal deletion of GATA4 gene locus was the most prominent feature across all liver cancer samples. Ectopic expression of GATA4 resulted in senescence of HCC cell lines. Mechanistically, GATA4 exerted tumor suppressive role by orchestrating the assembly of a tumor suppressor enhancing module: GATA4 directly bound and potently inhibited the mRNA transcription activity of β-catenin; meanwhile, β-catenin was recruited by GATA4 to promoter regions and facilitated transcription of GATA4 target genes, which were TSGs per se. Expression of GATA4 was effective to shrink GATA4-deficient HCC tumors in vivo. We also showed that β-catenin inhibitor was capable of shrinking GATA4-deficient tumors. Conclusions: Our study unveiled a previously unnoticed tumor suppressor enhancing module assembled by ectopically expressed GATA4 in HCC cells and denoted a therapeutic opportunity for GATA4 deficient HCC patients. Our study also presented an interesting case that an oncogenic transcription factor conditionally functioned as a tumor suppressor when recruited by a TSG transcription factor.
Keywords: GATA4; Hepatocellular carcinoma; Tumor suppressor gene; targeting therapy.
© The author(s).
Conflict of interest statement
Competing Interests: The authors have declared that no competing interest exists.
Figures
References
-
- Siegel RL, Miller KD, Jemal A. Cancer Statistics, 2017. CA Cancer J Clin. 2017;67:7–30. - PubMed
-
- Zhou M, Wang H, Zhu J, Chen W, Wang L, Liu S. et al. Cause-specific mortality for 240 causes in China during 1990-2013: a systematic subnational analysis for the Global Burden of Disease Study 2013. Lancet. 2016;387:251–72. - PubMed
-
- Kew MC. Epidemiology of hepatocellular carcinoma in sub-Saharan Africa. Ann Hepatol. 2013;12:173–82. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
