

Efficacy and safety of D,L-3-hydroxybutyrate (D,L-3-HB) treatment in multiple acyl-CoA dehydrogenase deficiency
- PMID: 31904027
- PMCID: PMC7200590
- DOI: 10.1038/s41436-019-0739-z
Efficacy and safety of D,L-3-hydroxybutyrate (D,L-3-HB) treatment in multiple acyl-CoA dehydrogenase deficiency
Abstract
Purpose: Multiple acyl-CoA dehydrogenase deficiency (MADD) is a life-threatening, ultrarare inborn error of metabolism. Case reports described successful D,L-3-hydroxybutyrate (D,L-3-HB) treatment in severely affected MADD patients, but systematic data on efficacy and safety is lacking.
Methods: A systematic literature review and an international, retrospective cohort study on clinical presentation, D,L-3-HB treatment method, and outcome in MADD(-like) patients.
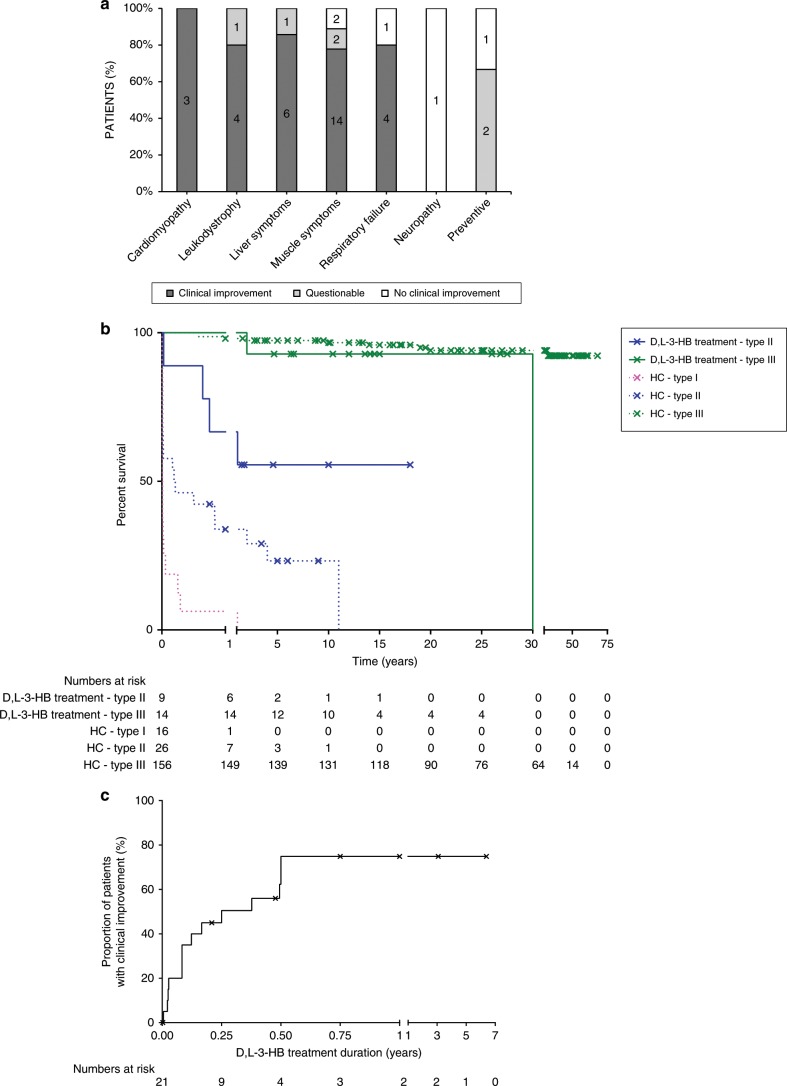
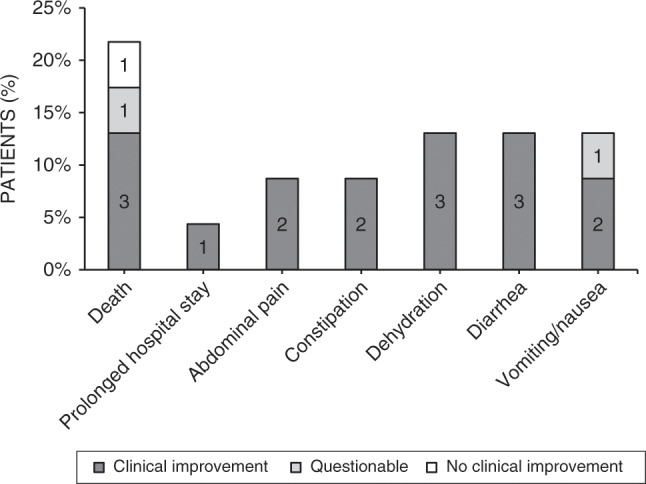
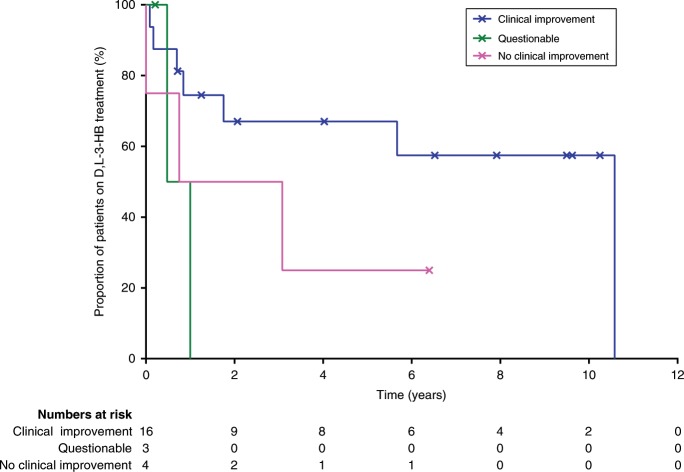
Results: Our study summarizes 23 MADD(-like) patients, including 14 new cases. Median age at clinical onset was two months (interquartile range [IQR]: 8 months). Median age at starting D,L-3-HB was seven months (IQR: 4.5 years). D,L-3-HB doses ranged between 100 and 2600 mg/kg/day. Clinical improvement was reported in 16 patients (70%) for cardiomyopathy, leukodystrophy, liver symptoms, muscle symptoms, and/or respiratory failure. D,L-3-HB appeared not effective for neuropathy. Survival appeared longer upon D,L-3-HB compared with historical controls. Median time until first clinical improvement was one month, and ranged up to six months. Reported side effects included abdominal pain, constipation, dehydration, diarrhea, and vomiting/nausea. Median D,L-3-HB treatment duration was two years (IQR: 6 years). D,L-3-HB treatment was discontinued in 12 patients (52%).
Conclusion: The strength of the current study is the international pooling of data demonstrating that D,L-3-HB treatment can be effective and safe in MADD(-like) patients.
Keywords: D,L-3-hydroxybutyrate treatment; fatty acid oxidation; inborn error of metabolism; ketone bodies; multiple acyl-CoA dehydrogenase deficiency.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
References
-
- Frerman FE, Goodman SI. Chapter 103: defects of electron transfer flavoprotein and electron transfer flavoprotein-ubiquinone oxidoreductase: glutaric acidemia type II. In: Valle D, Beaudet AL, Vogelstein B, editors. The online metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2014.
-
- Olpin SE. Implications of impaired ketogenesis in fatty acid oxidation disorders. Prostaglandins Leukot Essent Fatty Acids. 2004;70:293–308. - PubMed
-
- Bouteldja N, Andersen LT, Moller N, Gormsen LC. Using positron emission tomography to study human ketone body metabolism: a review. Metabolism. 2014;63:1375–1384. - PubMed
-
- Rinaldo P, Matern D, Bennett MJ. Fatty acid oxidation disorders. Annu Rev Physiol. 2002;64:477–502. - PubMed
-
- Bonham JR, Tanner MS, Pollitt RJ, et al. Oral sodium 3-hydroxybutyrate, a novel adjunct to treatment for multiple acyl CoA dehydrogenase deficiency. J Inherit Metab Dis. 1999;22(Suppl 1):101.
