

Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development
- PMID: 31904347
- PMCID: PMC7024565
- DOI: 10.1016/j.brainres.2019.146644
Sex differences in Mecp2-mutant Rett syndrome model mice and the impact of cellular mosaicism in phenotype development
Abstract
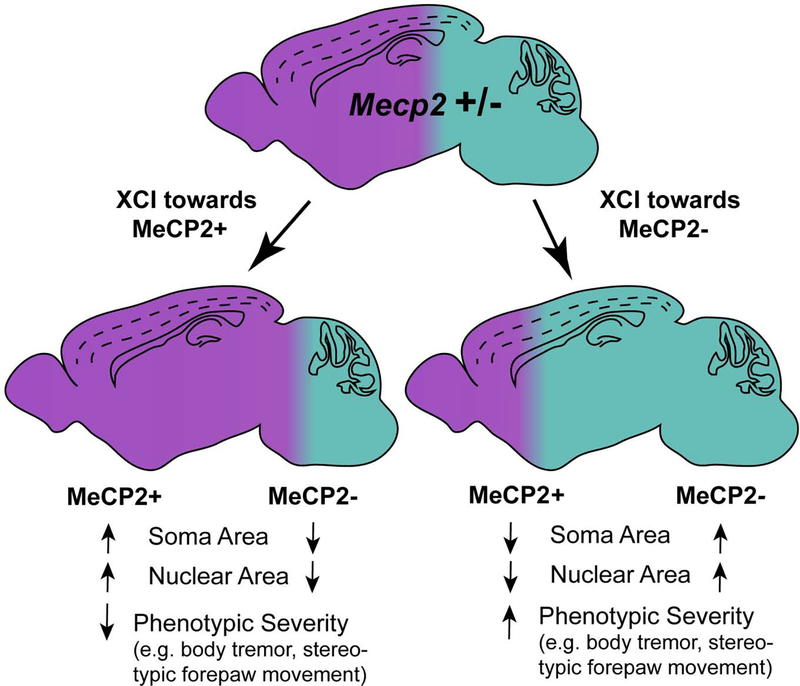
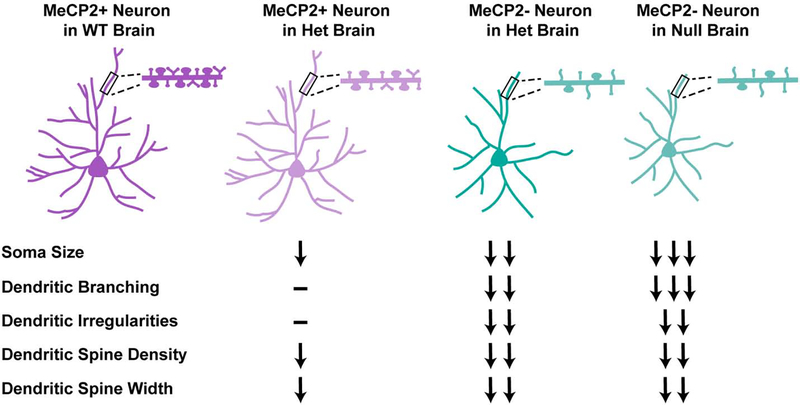
There is currently no effective treatment for Rett syndrome (RTT), a severe X-linked progressive neurodevelopmental disorder caused by mutations in the transcriptional regulator MECP2. Because MECP2 is subjected to X-inactivation, most affected individuals are female heterozygotes who display cellular mosaicism for normal and mutant MECP2. Males who are hemizygous for mutant MECP2 are more severely affected than heterozygous females and rarely survive. Mecp2 loss-of-function is less severe in mice, however, and male hemizygous null mice not only survive until adulthood, they have been the most commonly studied model system. Although heterozygous female mice better recapitulate human RTT, they have not been as thoroughly characterized. This is likely because of the added experimental challenges that they present, including delayed and more variable phenotypic progression and cellular mosaicism due to X-inactivation. In this review, we compare phenotypes of Mecp2 heterozygous female mice and male hemizygous null mouse models. Further, we discuss the complexities that arise from the many cell-type and tissue-type specific roles of MeCP2, as well as the combination of cell-autonomous and non-cell-autonomous disruptions that result from Mecp2 loss-of-function. This is of particular importance in the context of the female heterozygous brain, composed of a mixture of MeCP2+ and MeCP2- cells, the ratio of which can alter RTT phenotypes in the case of skewed X-inactivation. The goal of this review is to provide a clearer understanding of the pathophysiological differences between the mouse models, which is an essential consideration in the design of future pre-clinical studies.
Keywords: Epigenetics; MeCP2; Rett syndrome; Sex differences; X-chromosome inactivation.
Copyright © 2020 Elsevier B.V. All rights reserved.
Figures
References
-
- Amir RE, Van Den Veyver IB, Schultz R, Malicki DM, Tran CQ, Dahle EJ, Philippi A, Timar L, Percy AK, Motil KJ, Lichtarge O, Smith EOB, Glaze DG, Zoghbi HY, 2000. Influence of mutation type and X chromosome inactivation on Rett syndrome phenotypes. Ann. Neurol 47, 670–679. 10.1002/1531-8249(200005)47:5<670::AID-ANA20>3.0.CO;2-F - DOI - PubMed
-
- Amir RE, van den Veyver IB, Wan M, Tran CQ, Francke U, Zoghbi HY, 1999. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet 23, 185–188. - PubMed
-
- Asgarihafshejani A, Nashmi R, Delaney KR, 2019. Cell-Genotype Specific Effects of Mecp2 Mutation on Spontaneous and Nicotinic Acetylcholine Receptor-Evoked Currents in Medial Prefrontal Cortical Pyramidal Neurons in Female Rett Model Mice. Neuroscience 414, 141–153. 10.1016/j.neuroscience.2019.07.008 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
