

Machine and deep learning approaches for cancer drug repurposing
- PMID: 31904426
- PMCID: PMC7723306
- DOI: 10.1016/j.semcancer.2019.12.011
Machine and deep learning approaches for cancer drug repurposing
Abstract
Knowledge of the underpinnings of cancer initiation, progression and metastasis has increased exponentially in recent years. Advanced "omics" coupled with machine learning and artificial intelligence (deep learning) methods have helped elucidate targets and pathways critical to those processes that may be amenable to pharmacologic modulation. However, the current anti-cancer therapeutic armamentarium continues to lag behind. As the cost of developing a new drug remains prohibitively expensive, repurposing of existing approved and investigational drugs is sought after given known safety profiles and reduction in the cost barrier. Notably, successes in oncologic drug repurposing have been infrequent. Computational in-silico strategies have been developed to aid in modeling biological processes to find new disease-relevant targets and discovering novel drug-target and drug-phenotype associations. Machine and deep learning methods have especially enabled leaps in those successes. This review will discuss these methods as they pertain to cancer biology as well as immunomodulation for drug repurposing opportunities in oncologic diseases.
Keywords: Artificial intelligence; Deep learning; Drug discovery; Drug repurposing; Machine learning.
Copyright © 2019 Elsevier Ltd. All rights reserved.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
The authors declare that there are no conflicts of interest.
Figures
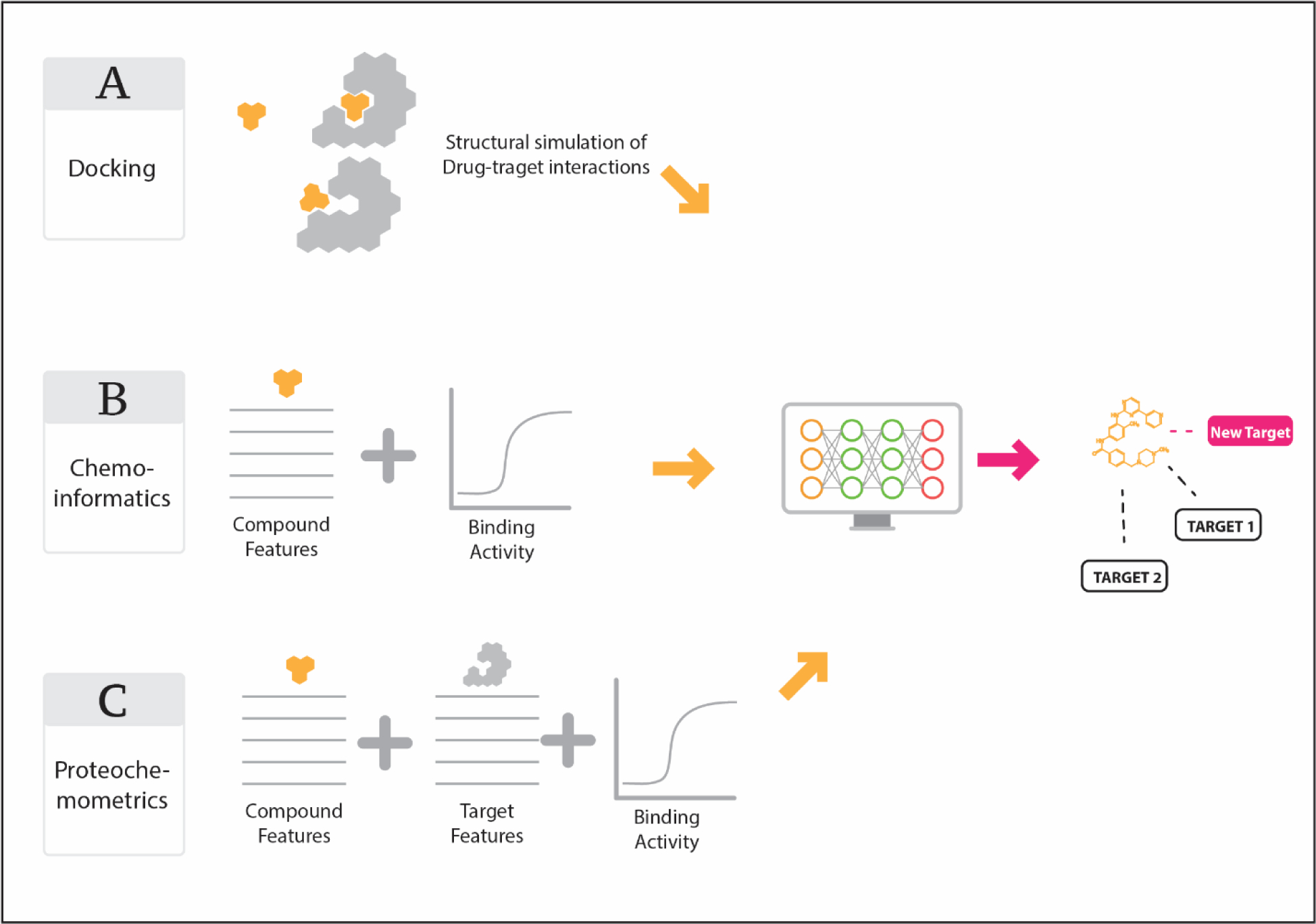
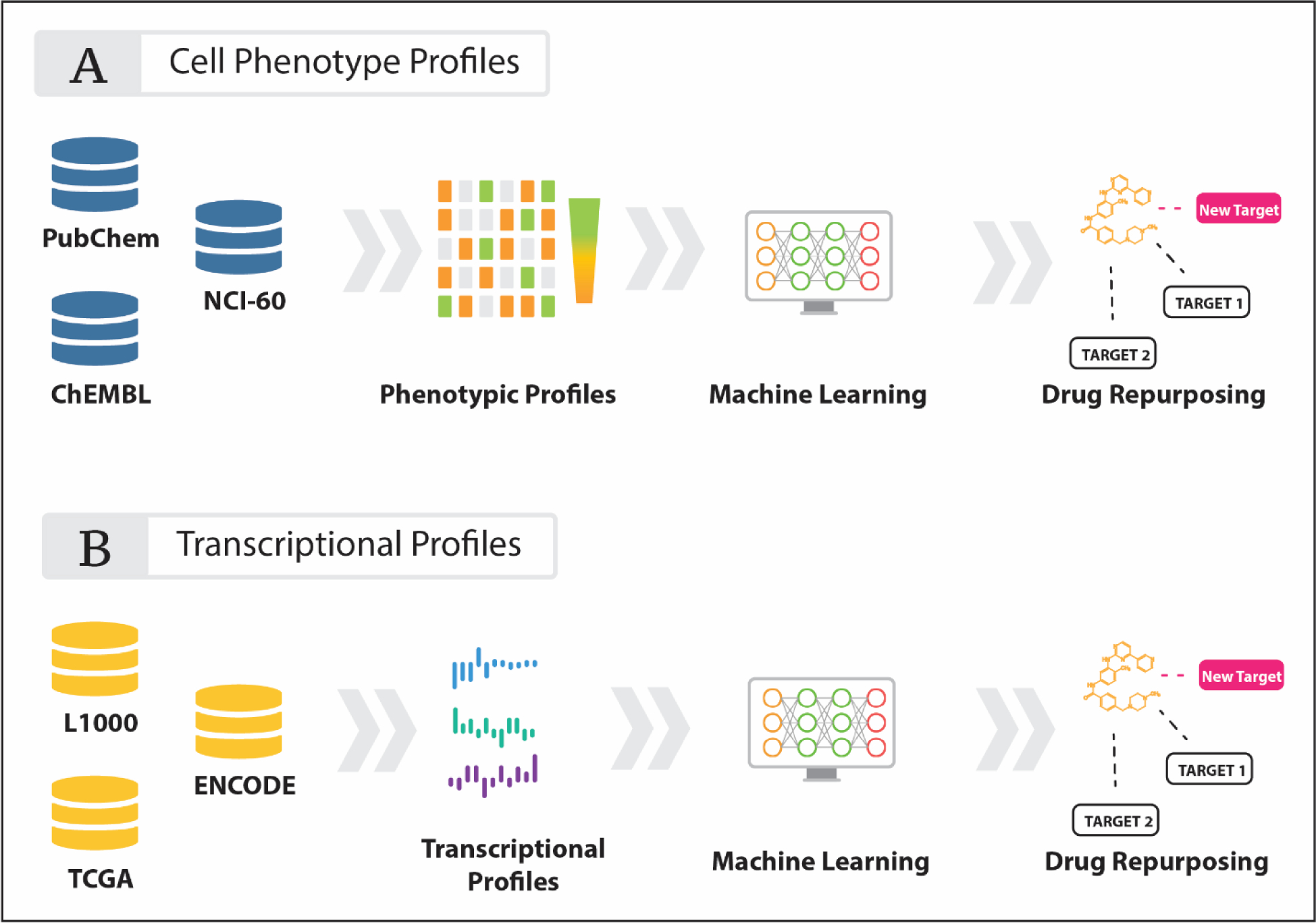
References
-
- Nosengo N Can you teach old drugs new tricks? Nature. 2016;534(7607):314–6. - PubMed
-
- Kitchen DB, Decornez H, Furr JR, Bajorath J. Docking and scoring in virtual screening for drug discovery: methods and applications. Nat Rev Drug Discov. 2004;3(11):935–49. - PubMed
-
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–49. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
