

Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates
- PMID: 31906474
- PMCID: PMC7017230
- DOI: 10.3390/genes11010050
Consensify: A Method for Generating Pseudohaploid Genome Sequences from Palaeogenomic Datasets with Reduced Error Rates
Abstract
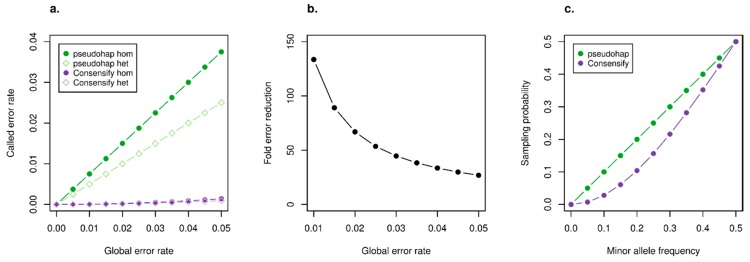
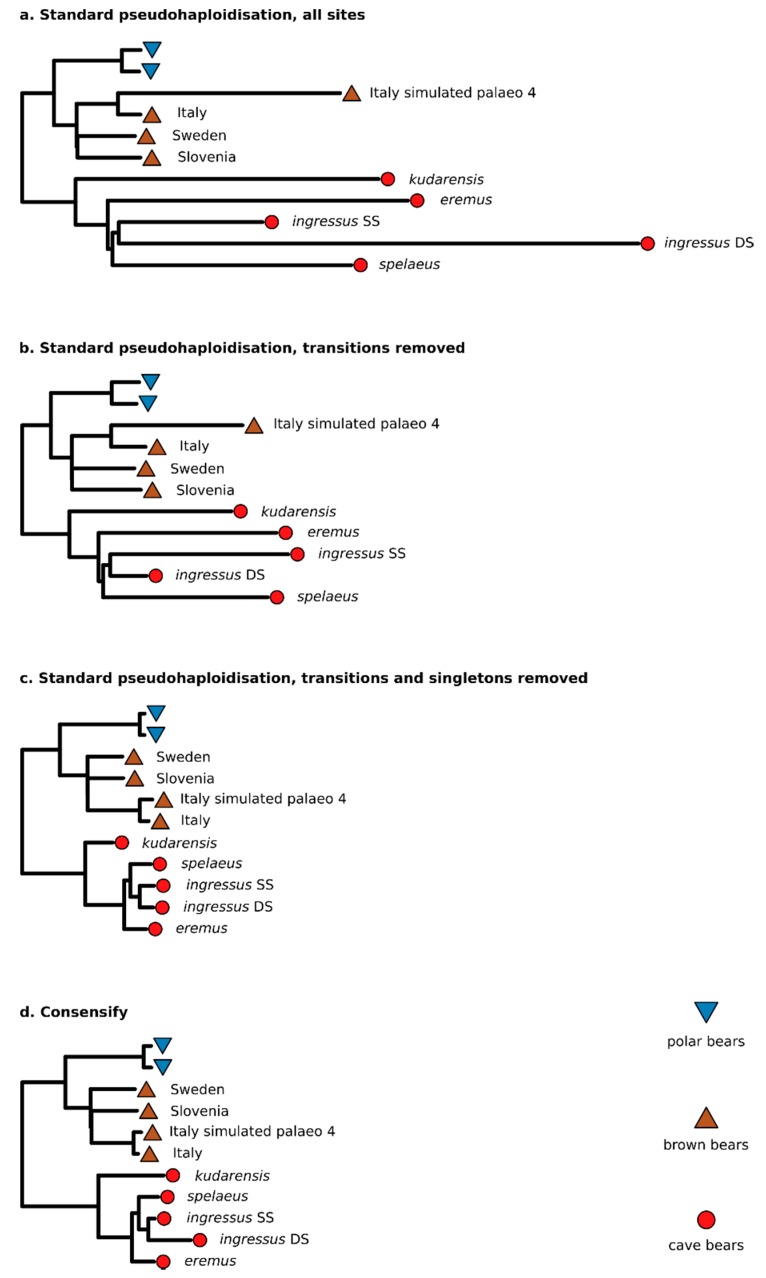
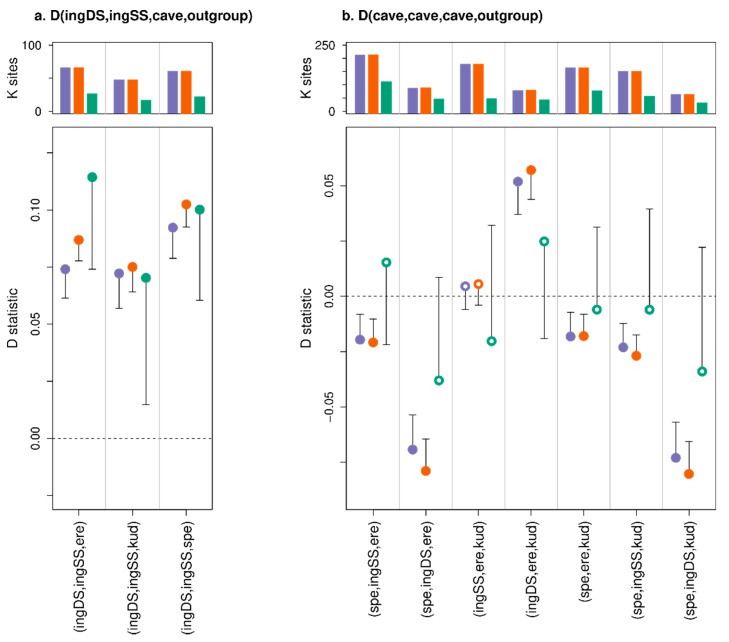
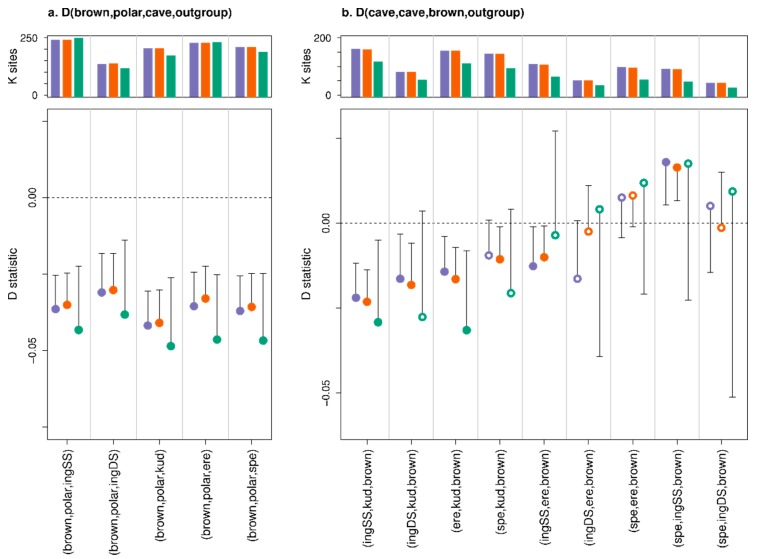
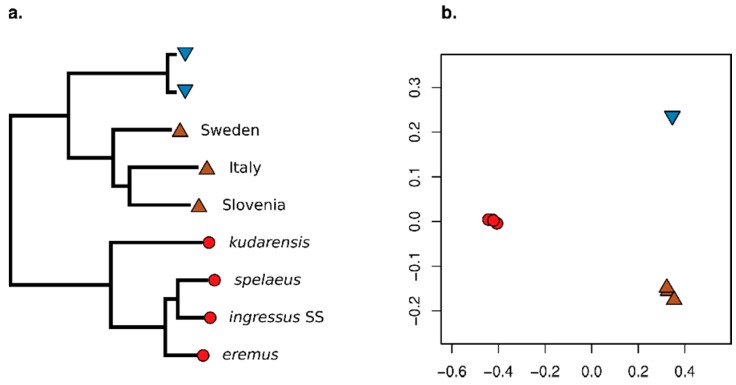
A standard practise in palaeogenome analysis is the conversion of mapped short read data into pseudohaploid sequences, frequently by selecting a single high-quality nucleotide at random from the stack of mapped reads. This controls for biases due to differential sequencing coverage, but it does not control for differential rates and types of sequencing error, which are frequently large and variable in datasets obtained from ancient samples. These errors have the potential to distort phylogenetic and population clustering analyses, and to mislead tests of admixture using D statistics. We introduce Consensify, a method for generating pseudohaploid sequences, which controls for biases resulting from differential sequencing coverage while greatly reducing error rates. The error correction is derived directly from the data itself, without the requirement for additional genomic resources or simplifying assumptions such as contemporaneous sampling. For phylogenetic and population clustering analysis, we find that Consensify is less affected by artefacts than methods based on single read sampling. For D statistics, Consensify is more resistant to false positives and appears to be less affected by biases resulting from different laboratory protocols than other frequently used methods. Although Consensify is developed with palaeogenomic data in mind, it is applicable for any low to medium coverage short read datasets. We predict that Consensify will be a useful tool for future studies of palaeogenomes.
Keywords: D statistics; ancient DNA; bioinformatics; error reduction; palaeogenomics; sequencing error.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Brotherton P., Endicott P., Sanchez J.J., Beaumont M., Barnett R., Austin J., Cooper A. Novel high-resolution characterization of ancient DNA reveals C > U-type base modification events as the sole cause of post mortem miscoding lesions. Nucleic Acids Res. 2007;35:5717–5728. doi: 10.1093/nar/gkm588. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
