

Biophysical prediction of protein-peptide interactions and signaling networks using machine learning
- PMID: 31907444
- PMCID: PMC7004877
- DOI: 10.1038/s41592-019-0687-1
Biophysical prediction of protein-peptide interactions and signaling networks using machine learning
Abstract
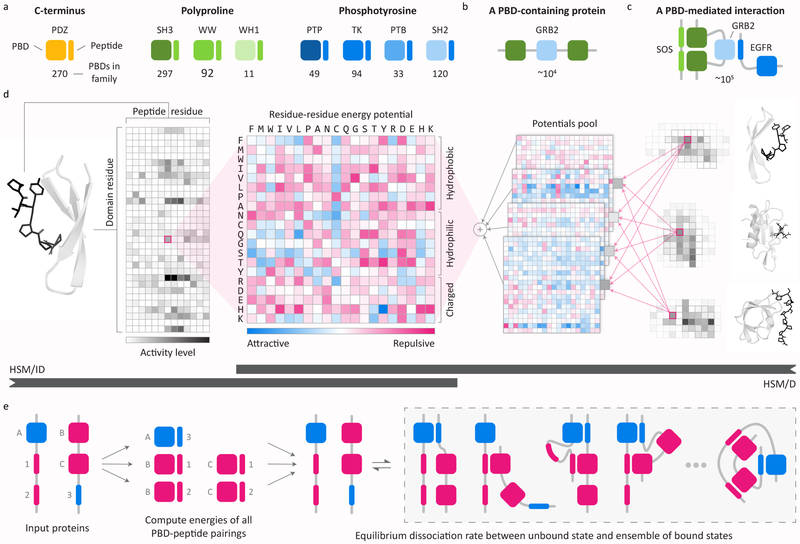
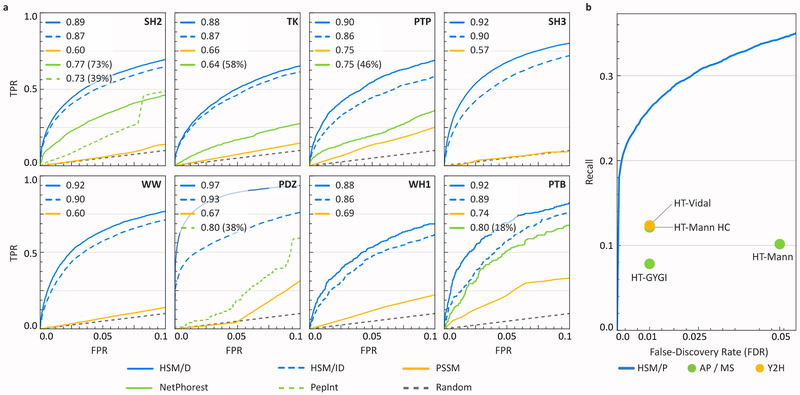
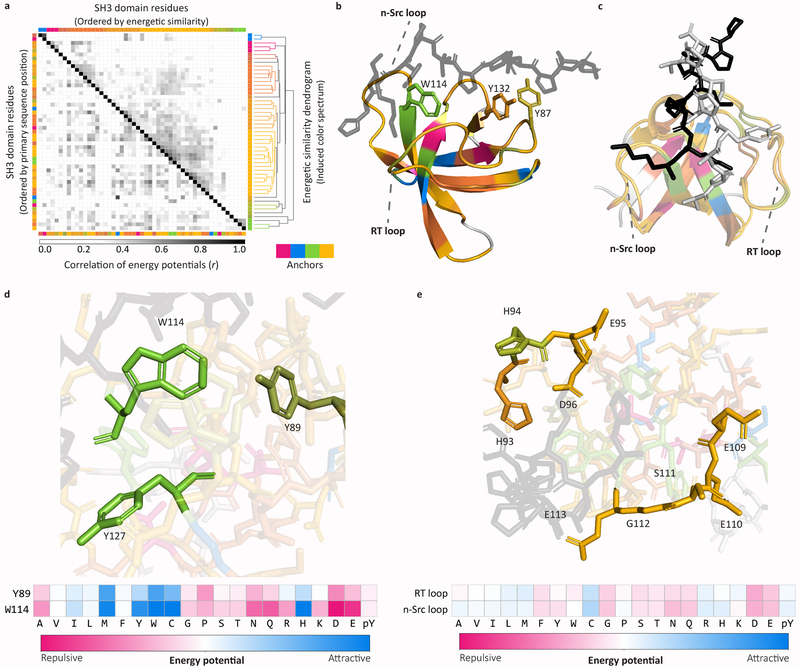
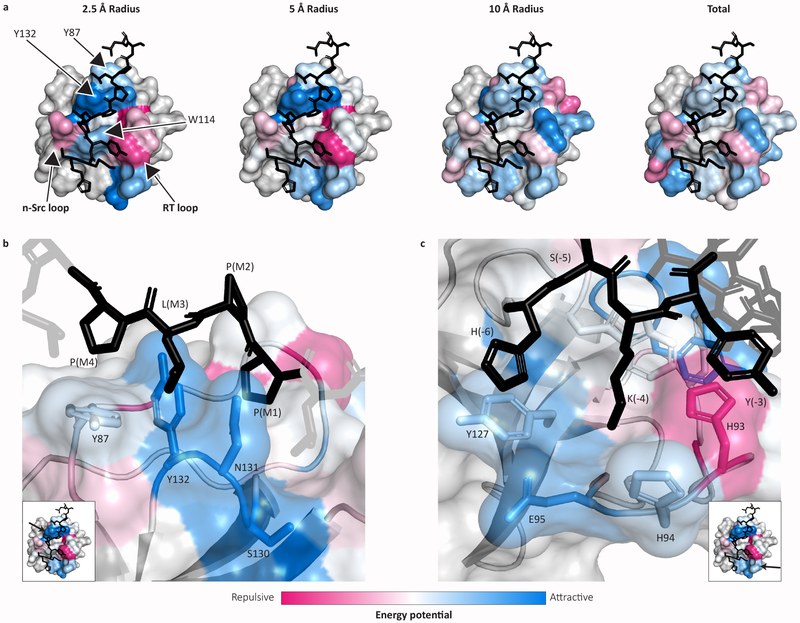
In mammalian cells, much of signal transduction is mediated by weak protein-protein interactions between globular peptide-binding domains (PBDs) and unstructured peptidic motifs in partner proteins. The number and diversity of these PBDs (over 1,800 are known), their low binding affinities and the sensitivity of binding properties to minor sequence variation represent a substantial challenge to experimental and computational analysis of PBD specificity and the networks PBDs create. Here, we introduce a bespoke machine-learning approach, hierarchical statistical mechanical modeling (HSM), capable of accurately predicting the affinities of PBD-peptide interactions across multiple protein families. By synthesizing biophysical priors within a modern machine-learning framework, HSM outperforms existing computational methods and high-throughput experimental assays. HSM models are interpretable in familiar biophysical terms at three spatial scales: the energetics of protein-peptide binding, the multidentate organization of protein-protein interactions and the global architecture of signaling networks.
Conflict of interest statement
COMPETING INTERESTS
PKS is a member of the SAB or Board of Directors of Merrimack Pharmaceutical, Glencoe Software, Applied Biomath and RareCyte Inc. and has equity in these companies; Sorger declares that none of these relationships are directly or indirectly related to the content of this manuscript.
Figures

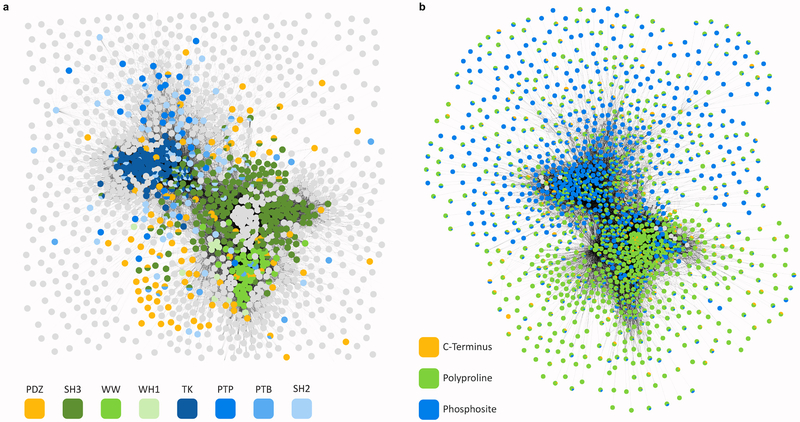
References
-
- Perkins JR, Diboun I, Dessailly BH, Lees JG & Orengo C Transient Protein-Protein Interactions: Structural, Functional, and Network Properties. Structure 18, 1233–1243 (2010). - PubMed
-
- Mayer BJ The discovery of modular binding domains: building blocks of cell signalling. Nat. Rev. Mol. Cell Biol 16, 691–698 (2015). - PubMed
-
- Tompa P, Davey NE, Gibson TJ & Babu MM A Million Peptide Motifs for the Molecular Biologist. Mol. Cell 55, 161–169 (2014). - PubMed
References (Online Methods-only)
-
- Sokal RR & Michener CD A Statistical Methods for Evaluating Relationships. Univ. Kans. Sci. Bull 38, 1409–1448.
-
- DeLong ER, DeLong DM & Clarke-Pearson DL Comparing the areas under two or more correlated receiver operating characteristic curves: a nonparametric approach. Biometrics 44, 837–845 (1988). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
