

Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data
- PMID: 31907445
- PMCID: PMC7098173
- DOI: 10.1038/s41592-019-0690-6
Benchmarking algorithms for gene regulatory network inference from single-cell transcriptomic data
Abstract
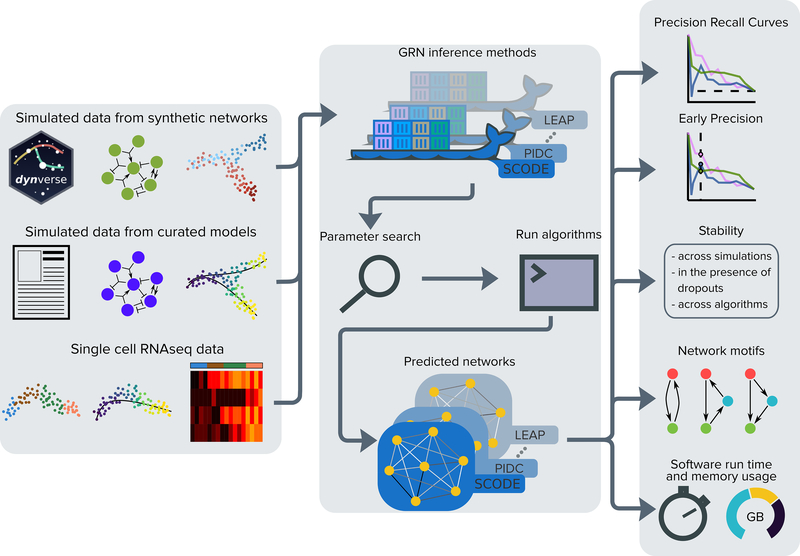
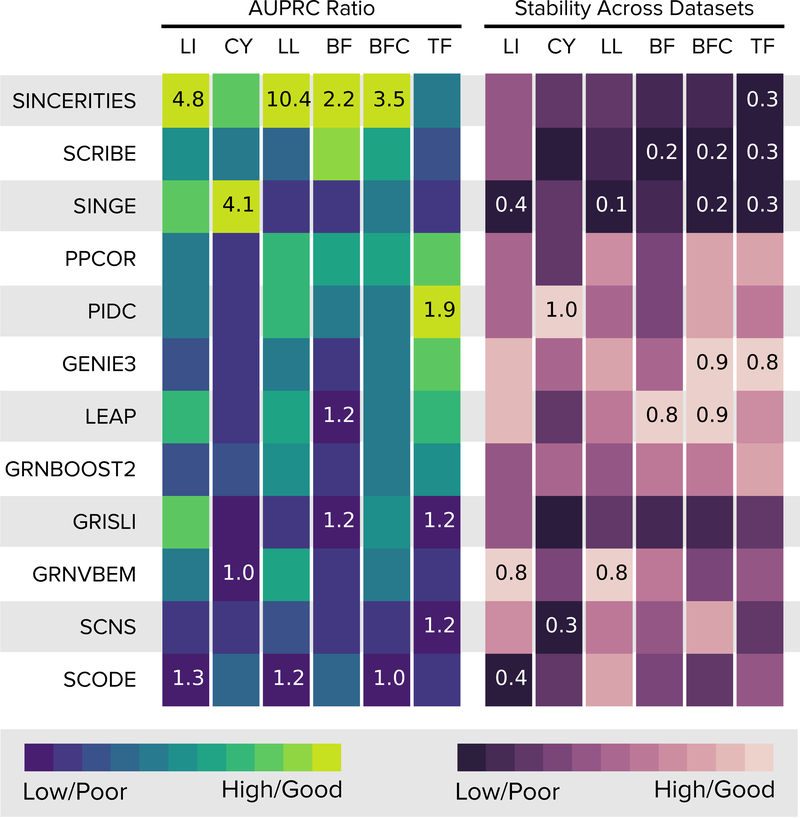
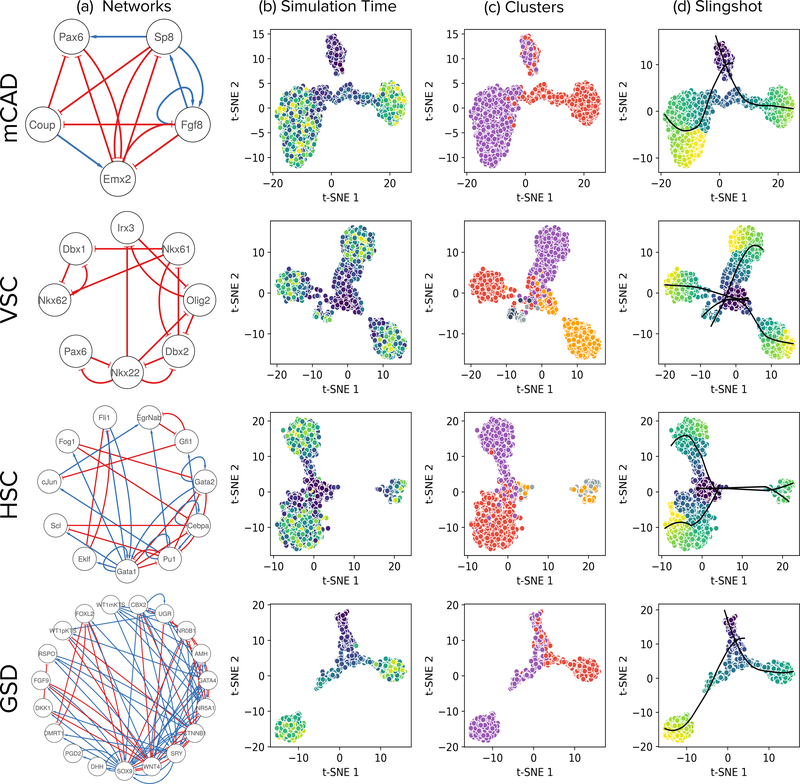
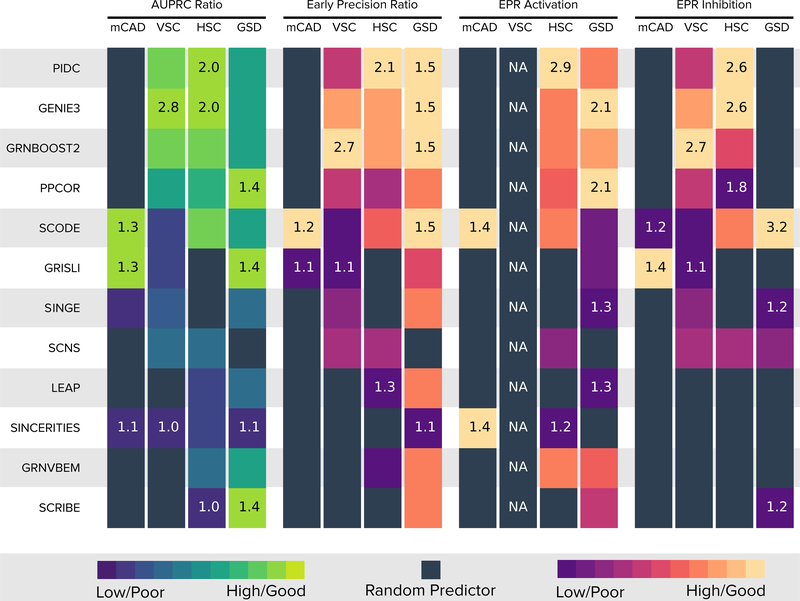
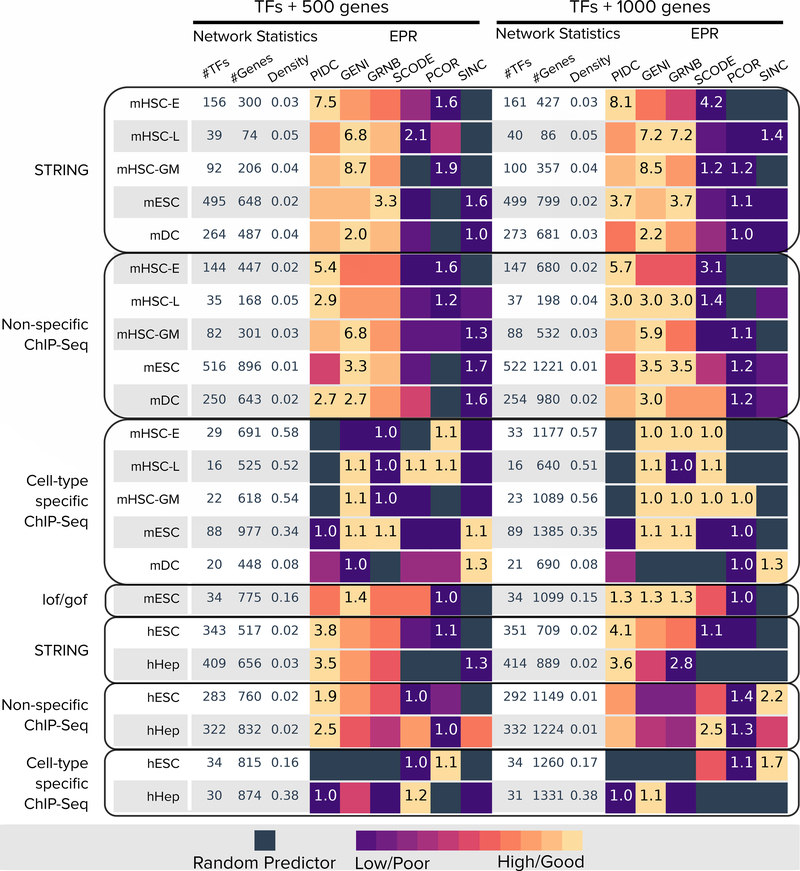
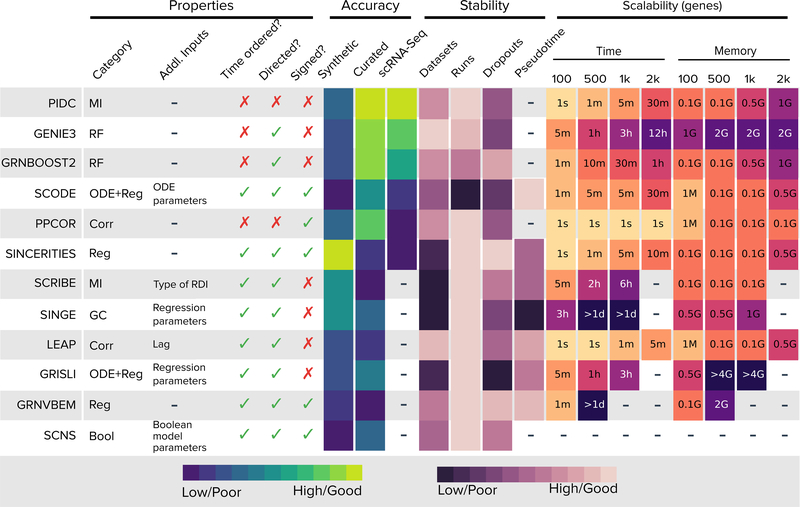
We present a systematic evaluation of state-of-the-art algorithms for inferring gene regulatory networks from single-cell transcriptional data. As the ground truth for assessing accuracy, we use synthetic networks with predictable trajectories, literature-curated Boolean models and diverse transcriptional regulatory networks. We develop a strategy to simulate single-cell transcriptional data from synthetic and Boolean networks that avoids pitfalls of previously used methods. Furthermore, we collect networks from multiple experimental single-cell RNA-seq datasets. We develop an evaluation framework called BEELINE. We find that the area under the precision-recall curve and early precision of the algorithms are moderate. The methods are better in recovering interactions in synthetic networks than Boolean models. The algorithms with the best early precision values for Boolean models also perform well on experimental datasets. Techniques that do not require pseudotime-ordered cells are generally more accurate. Based on these results, we present recommendations to end users. BEELINE will aid the development of gene regulatory network inference algorithms.
Conflict of interest statement
Competing Interests
The authors declare no competing interests.
Figures
References
-
- Rozenblatt-Rosen O, Stubbington MJT, Regev A & Teichmann SA The Human Cell Atlas: from vision to reality. Nature 550, 451–453 (2017). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
