

High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis
- PMID: 31907459
- PMCID: PMC7005909
- DOI: 10.1038/s41591-019-0709-7
High-resolution mycobiota analysis reveals dynamic intestinal translocation preceding invasive candidiasis
Abstract
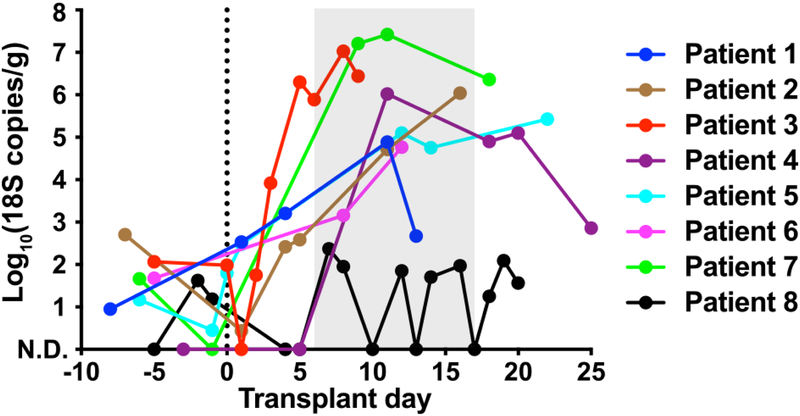
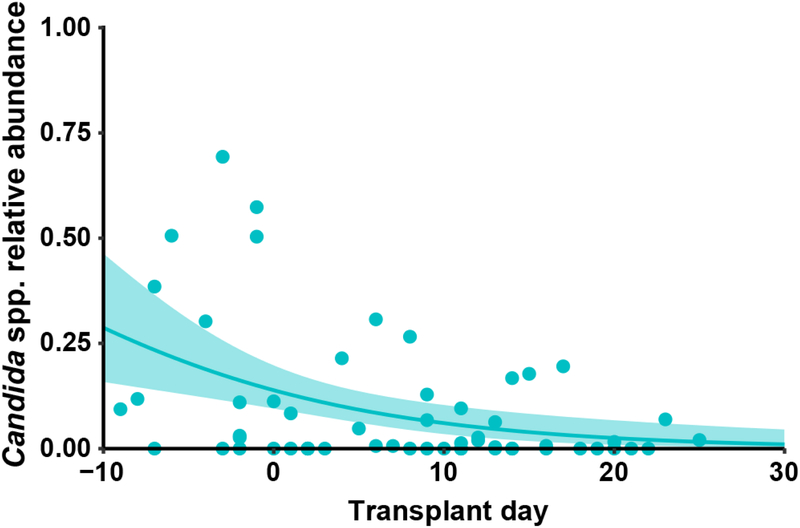
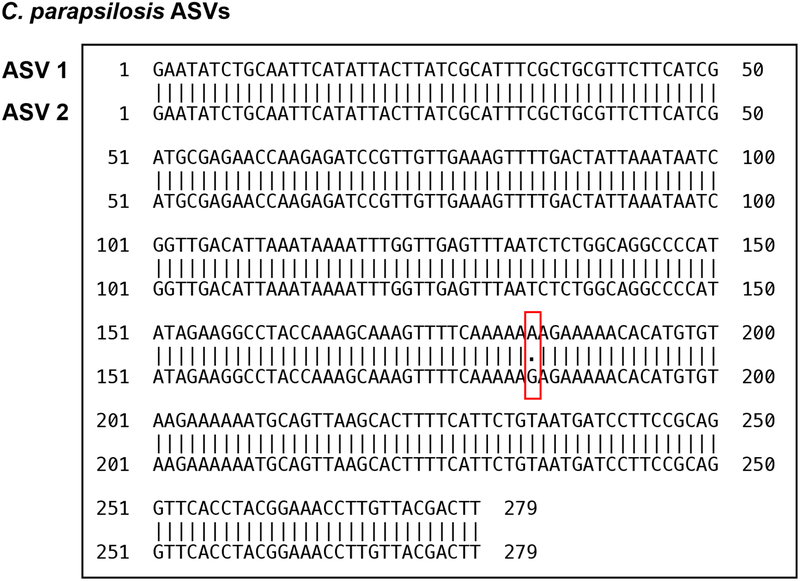
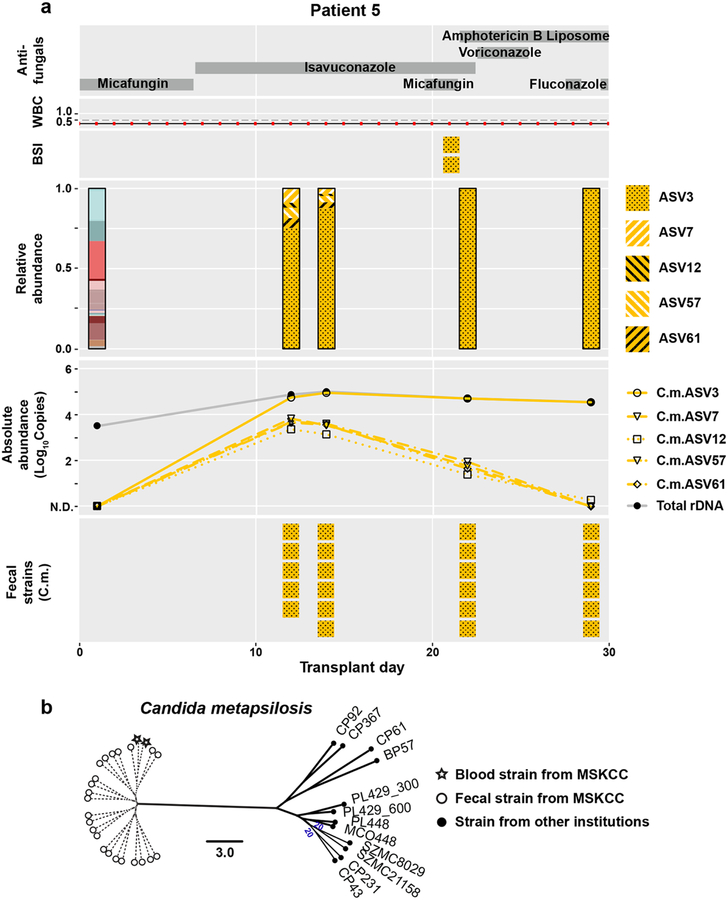
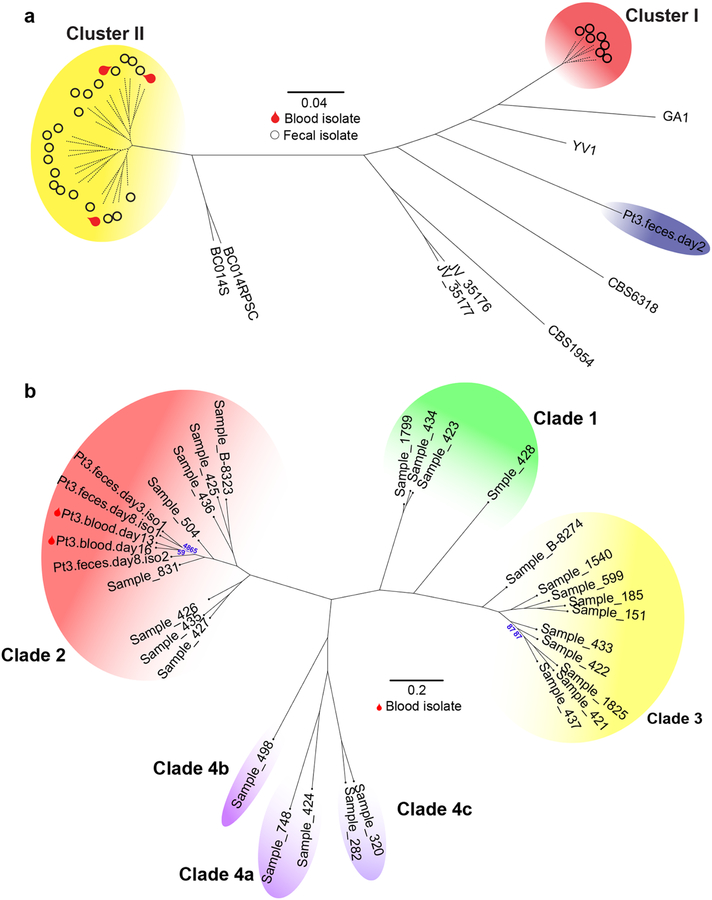
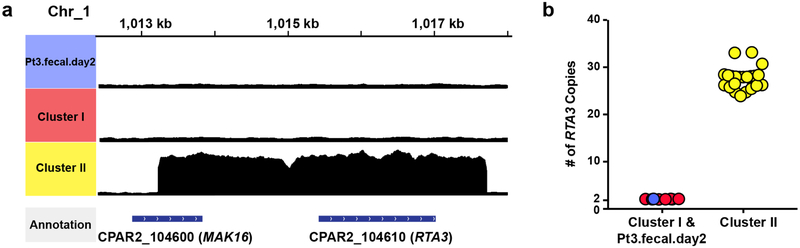
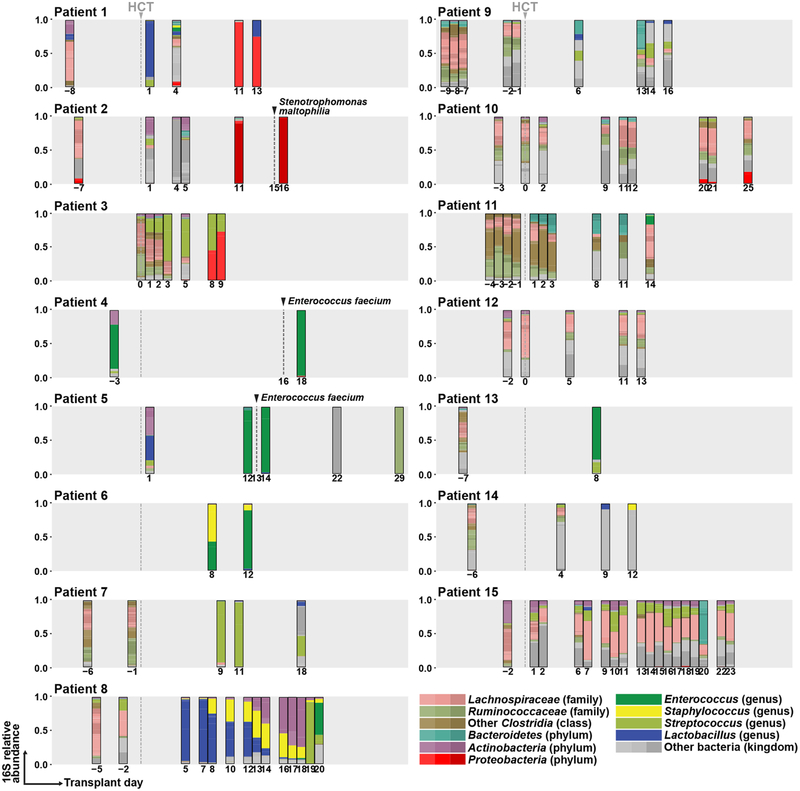
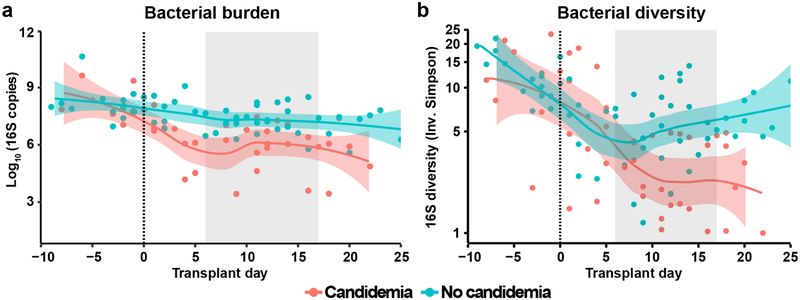
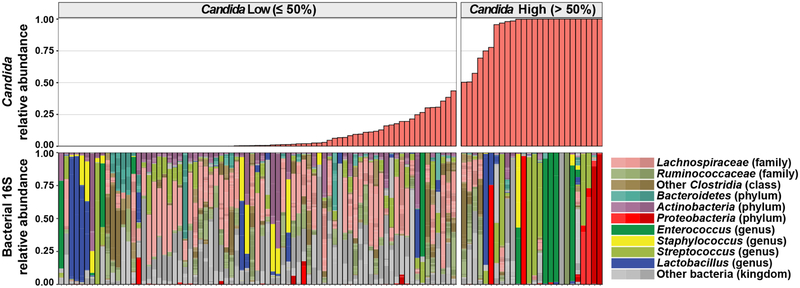
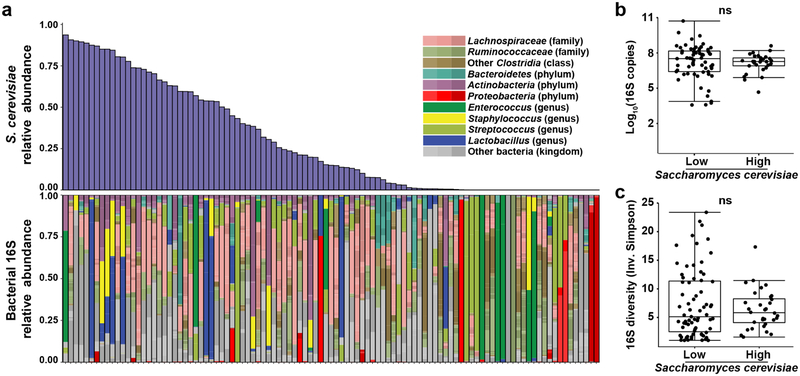
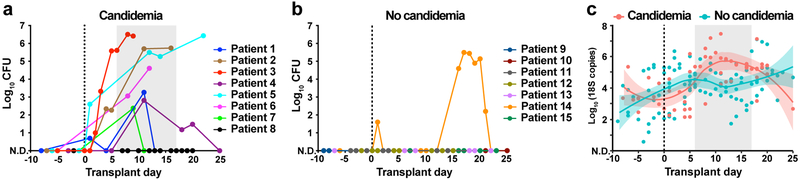
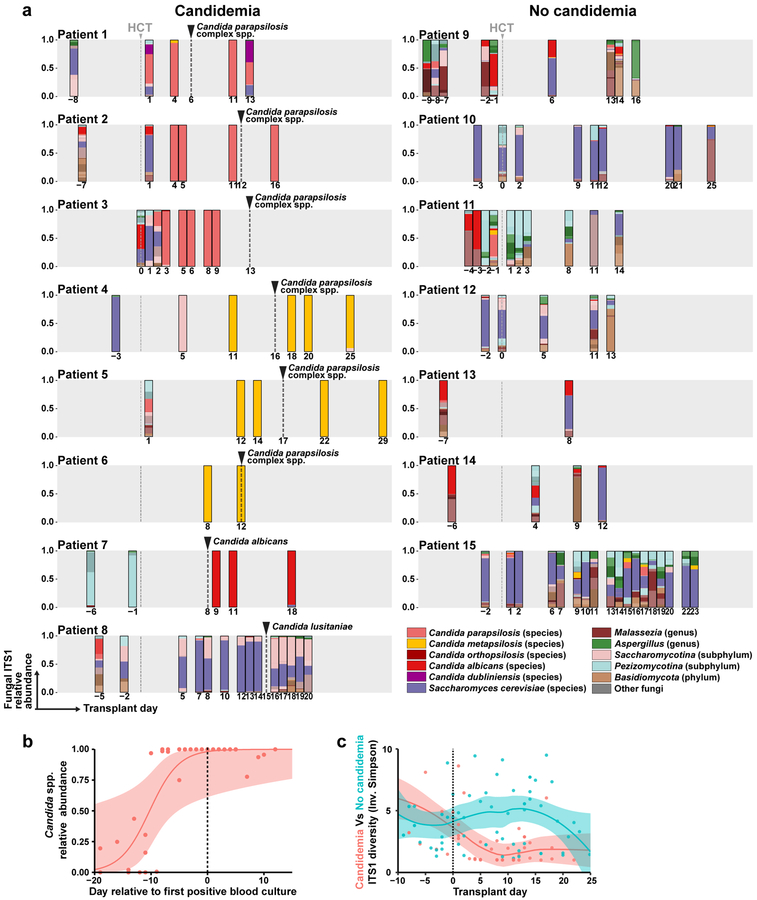
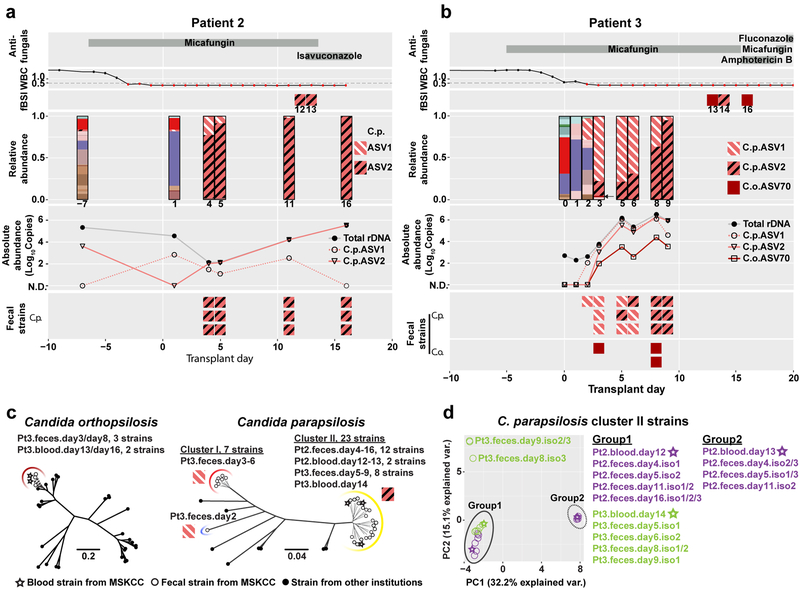
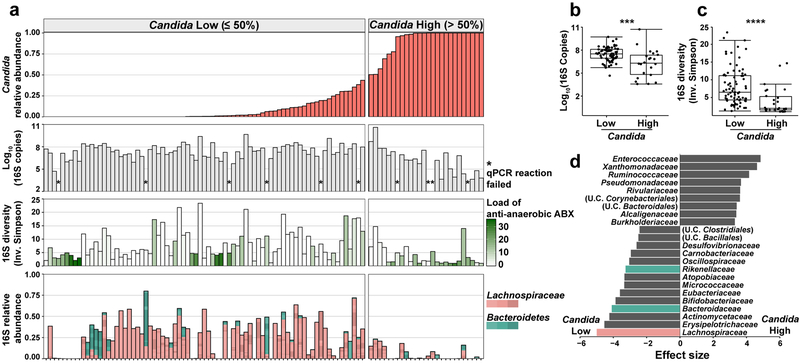
The intestinal microbiota is a complex community of bacteria, archaea, viruses, protists and fungi1,2. Although the composition of bacterial constituents has been linked to immune homeostasis and infectious susceptibility3-7, the role of non-bacterial constituents and cross-kingdom microbial interactions in these processes is poorly understood2,8. Fungi represent a major cause of infectious morbidity and mortality in immunocompromised individuals, although the relationship of intestinal fungi (that is, the mycobiota) with fungal bloodstream infections remains undefined9. We integrated an optimized bioinformatics pipeline with high-resolution mycobiota sequencing and comparative genomic analyses of fecal and blood specimens from recipients of allogeneic hematopoietic cell transplant. Patients with Candida bloodstream infection experienced a prior marked intestinal expansion of pathogenic Candida species; this expansion consisted of a complex dynamic between multiple species and subspecies with a stochastic translocation pattern into the bloodstream. The intestinal expansion of pathogenic Candida spp. was associated with a substantial loss in bacterial burden and diversity, particularly in the anaerobes. Thus, simultaneous analysis of intestinal fungi and bacteria identifies dysbiosis states across kingdoms that may promote fungal translocation and facilitate invasive disease. These findings support microbiota-driven approaches to identify patients at risk of fungal bloodstream infections for pre-emptive therapeutic intervention.
Conflict of interest statement
Figures
Comment in
-
Tracing the Origin of Invasive Fungal Infections.Trends Microbiol. 2020 Apr;28(4):240-242. doi: 10.1016/j.tim.2020.01.007. Epub 2020 Feb 7. Trends Microbiol. 2020. PMID: 32044130
References
-
- Richard ML & Sokol H The gut mycobiota: insights into analysis, environmental interactions and role in gastrointestinal diseases. Nat Rev Gastroenterol Hepatol 16, 331–345 (2019). - PubMed
-
- Lewis BB & Pamer EG Microbiota-Based Therapies for Clostridium difficile and Antibiotic-Resistant Enteric Infections. Annu Rev Microbiol 71, 157–178 (2017). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
