

Future treatments for hereditary hemorrhagic telangiectasia
- PMID: 31910860
- PMCID: PMC6945546
- DOI: 10.1186/s13023-019-1281-4
Future treatments for hereditary hemorrhagic telangiectasia
Abstract
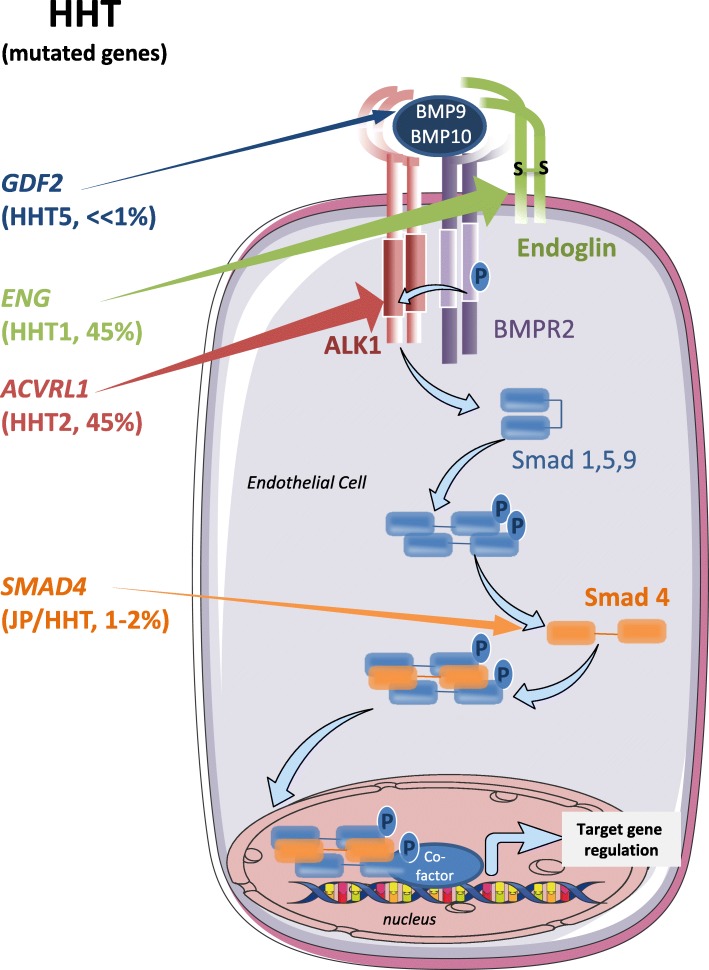
Hereditary Hemorrhagic Telangiectasia (HHT), also known as Rendu-Osler syndrome, is a genetic vascular disorder affecting 1 in 5000-8000 individuals worldwide. This rare disease is characterized by various vascular defects including epistaxis, blood vessel dilations (telangiectasia) and arteriovenous malformations (AVM) in several organs. About 90% of the cases are associated with heterozygous mutations of ACVRL1 or ENG genes, that respectively encode a bone morphogenetic protein receptor (activin receptor-like kinase 1, ALK1) and a co-receptor named endoglin. Less frequent mutations found in the remaining 10% of patients also affect the gene SMAD4 which is part of the transcriptional complex directly activated by this pathway. Presently, the therapeutic treatments for HHT are intended to reduce the symptoms of the disease. However, recent progress has been made using drugs that target VEGF (vascular endothelial growth factor) and the angiogenic pathway with the use of bevacizumab (anti-VEGF antibody). Furthermore, several exciting high-throughput screenings and preclinical studies have identified new molecular targets directly related to the signaling pathways affected in the disease. These include FKBP12, PI3-kinase and angiopoietin-2. This review aims at reporting these recent developments that should soon allow a better care of HHT patients.
Keywords: ALK1; Bevacizumab; Bone morphogenetic protein signaling; Drug repositioning; Hereditary hemorrhagic telangiectasia; High throughput screening; Tacrolimus; Vascular malformations.
Conflict of interest statement
The authors declare that they have no competing interest.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
