

Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines
- PMID: 31911591
- PMCID: PMC6946708
- DOI: 10.1038/s41467-019-13809-8
Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines
Erratum in
-
Author Correction: Ketamine disinhibits dendrites and enhances calcium signals in prefrontal dendritic spines.Nat Commun. 2021 Jan 8;12(1):370. doi: 10.1038/s41467-020-20634-x. Nat Commun. 2021. PMID: 33420087 Free PMC article. No abstract available.
Abstract
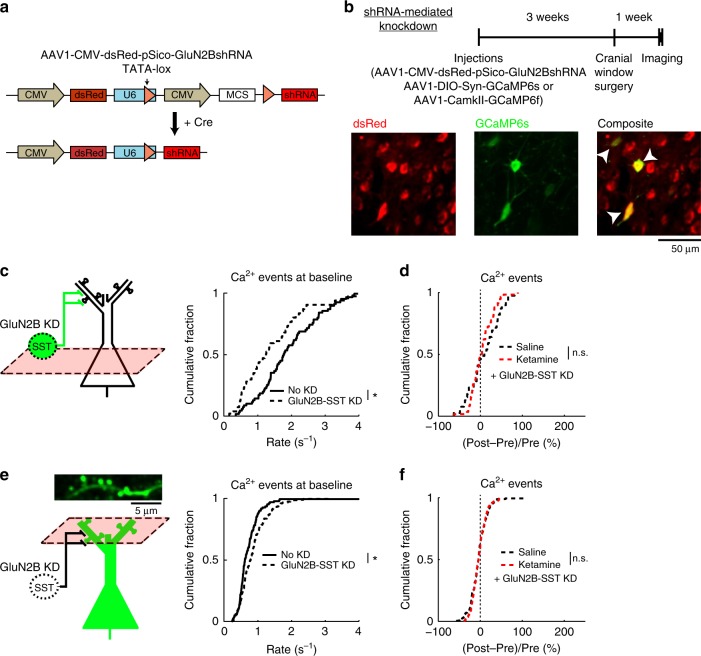
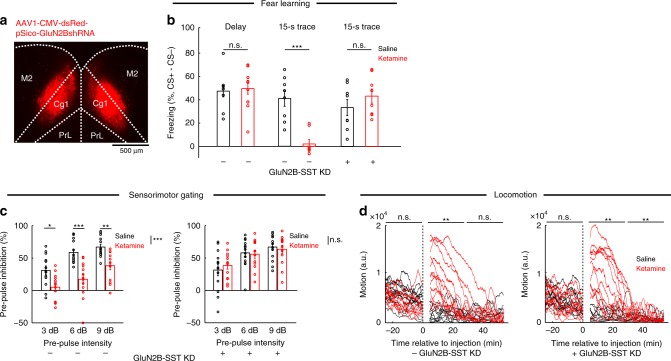
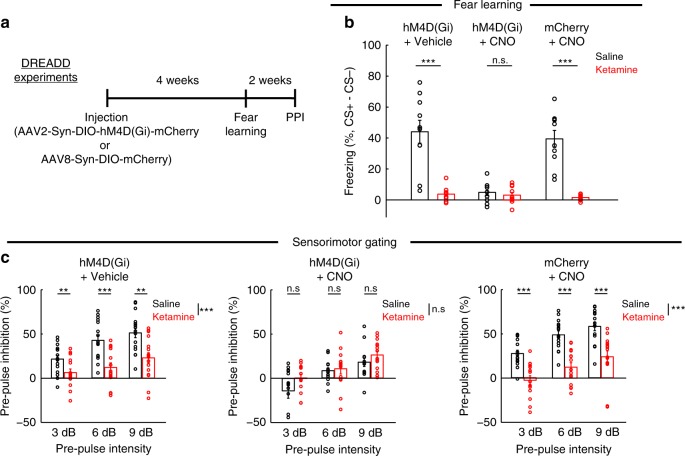
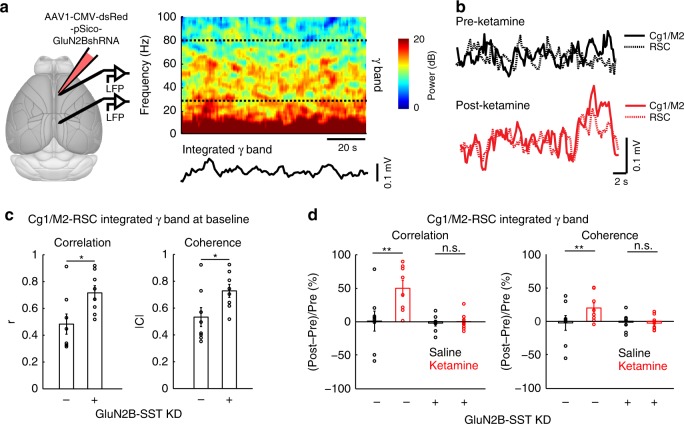
A subanesthetic dose of ketamine causes acute psychotomimetic symptoms and sustained antidepressant effects. In prefrontal cortex, the prevailing disinhibition hypothesis posits that N-methyl-d-aspartate receptor (NMDAR) antagonists such as ketamine act preferentially on GABAergic neurons. However, cortical interneurons are heterogeneous. In particular, somatostatin-expressing (SST) interneurons selectively inhibit dendrites and regulate synaptic inputs, yet their response to systemic NMDAR antagonism is unknown. Here, we report that ketamine acutely suppresses the activity of SST interneurons in the medial prefrontal cortex of the awake mouse. The deficient dendritic inhibition leads to greater synaptically evoked calcium transients in the apical dendritic spines of pyramidal neurons. By manipulating NMDAR signaling via GluN2B knockdown, we show that ketamine's actions on the dendritic inhibitory mechanism has ramifications for frontal cortex-dependent behaviors and cortico-cortical connectivity. Collectively, these results demonstrate dendritic disinhibition and elevated calcium levels in dendritic spines as important local-circuit alterations driven by the administration of subanesthetic ketamine.
Conflict of interest statement
R.S.D. has consulted and/or received research support from Allergan, Naurex, Aptynix, Lilly, Johnson & Johnson, Taisho, Relmada, and Navitor on unrelated projects. The remaining authors declare no competing interests.
Figures
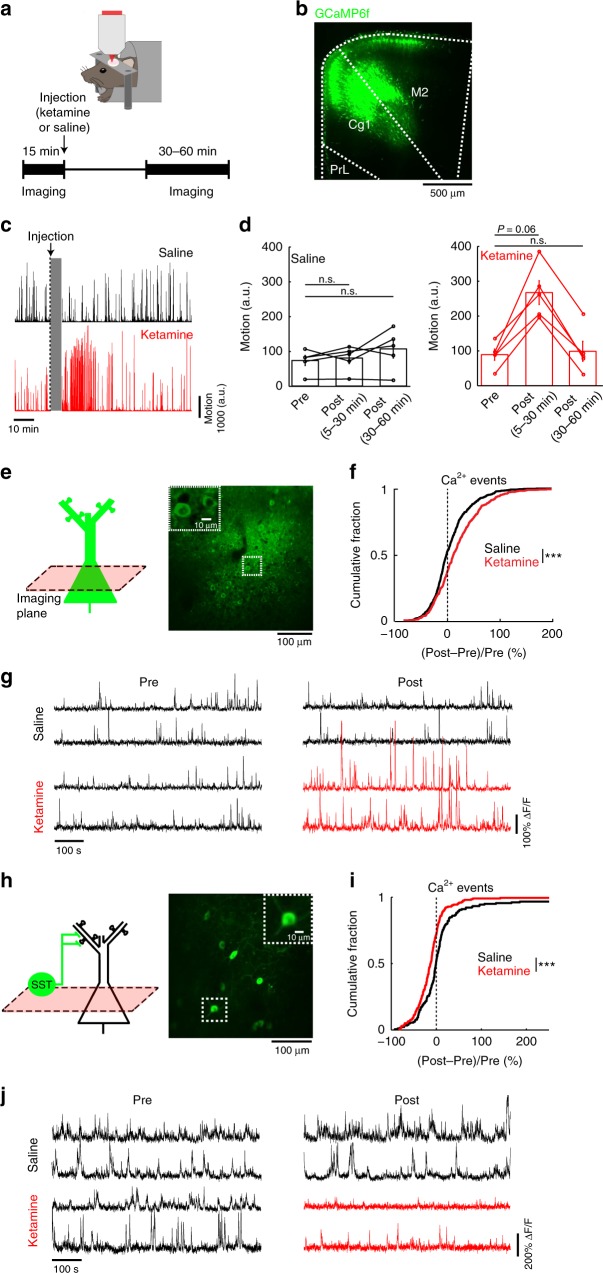
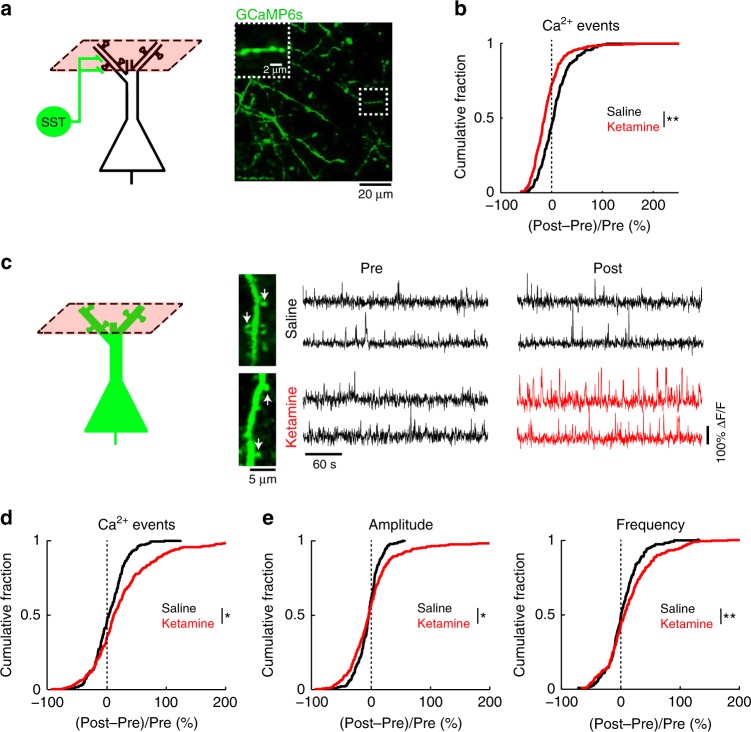
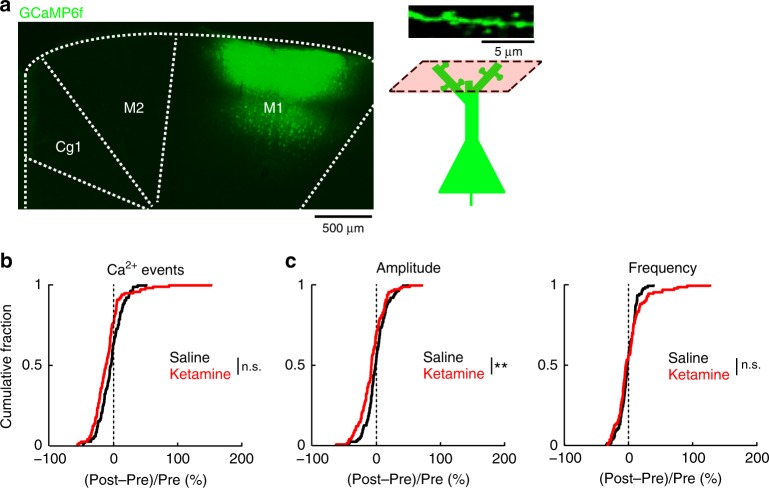
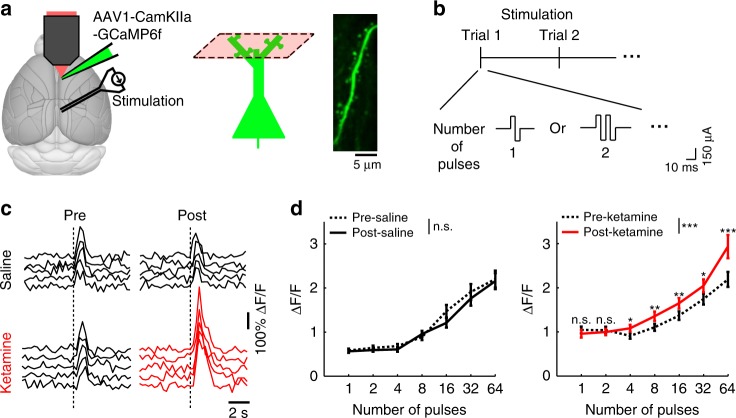
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
