

A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia
- PMID: 31913266
- PMCID: PMC6949222
- DOI: 10.1038/s41408-019-0270-0
A new regulatory mechanism of protein phosphatase 2A activity via SET in acute myeloid leukemia
Abstract
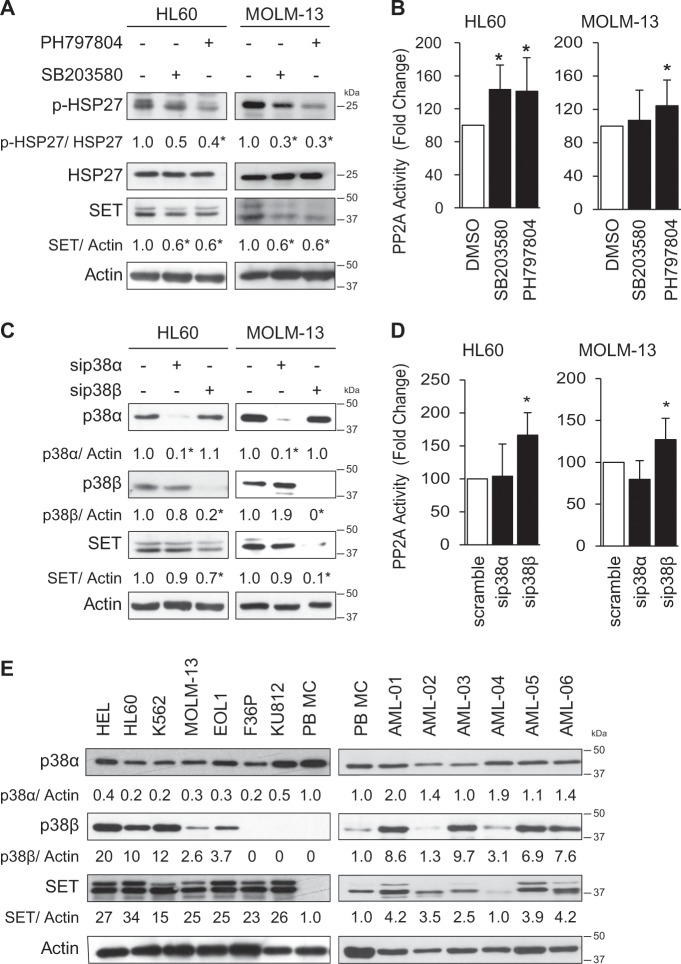
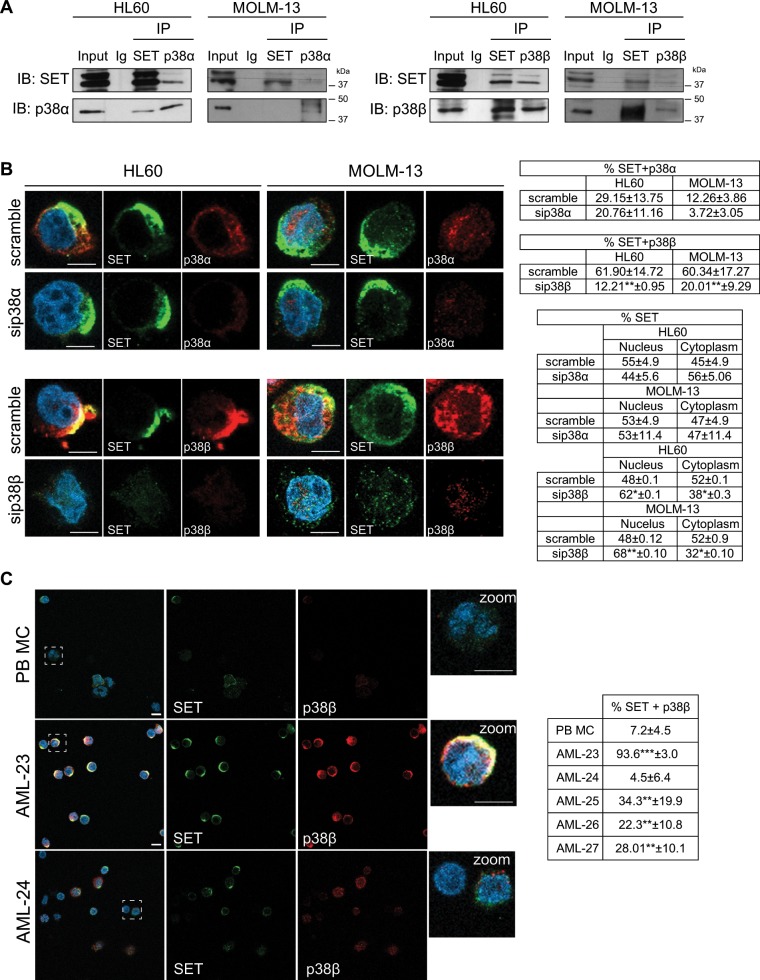
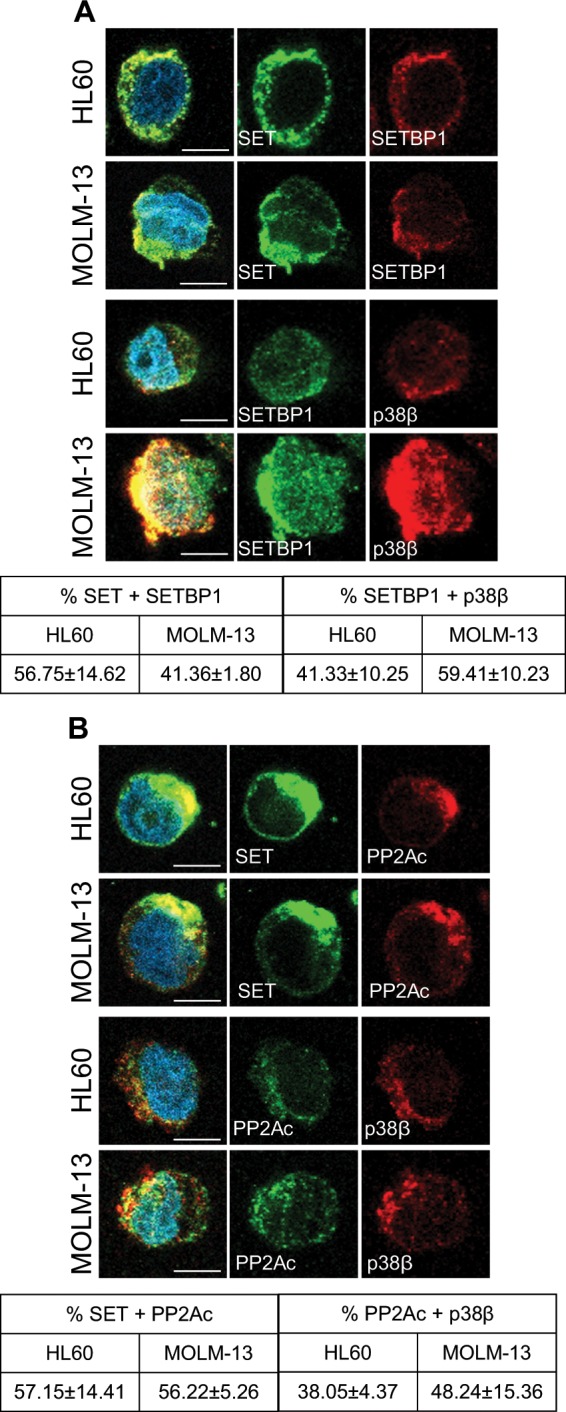
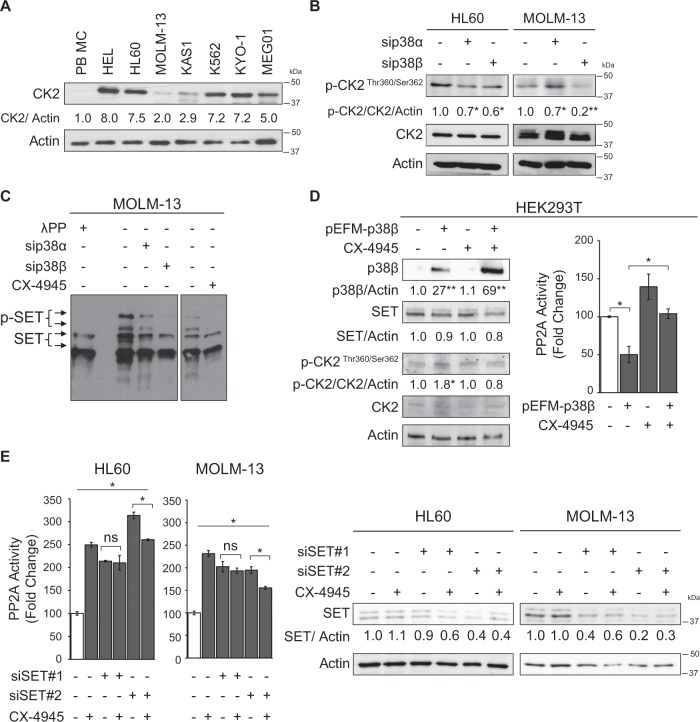
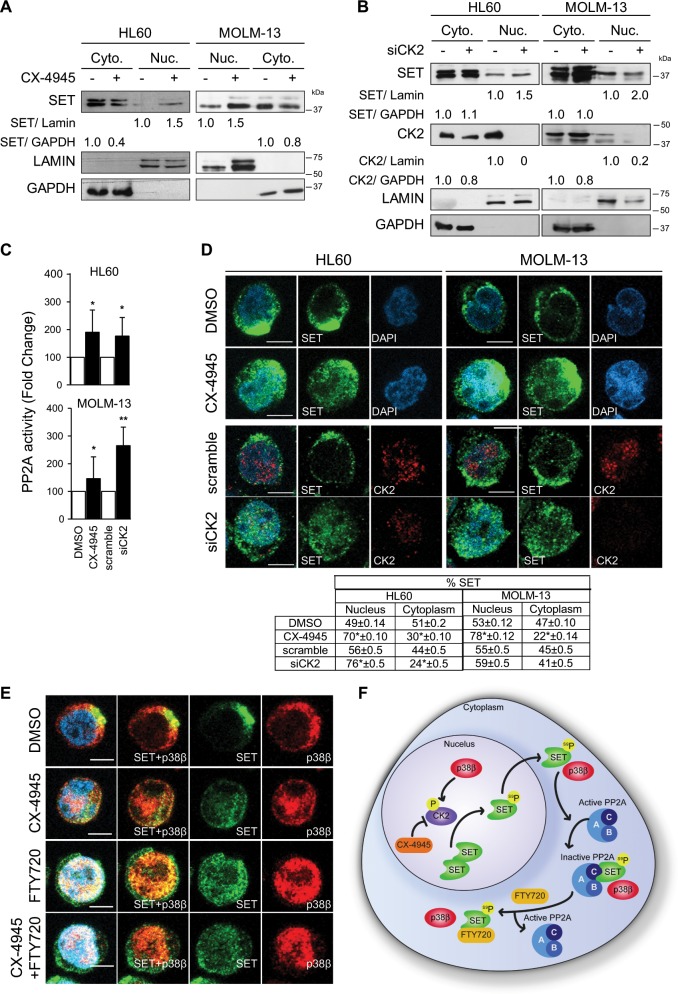
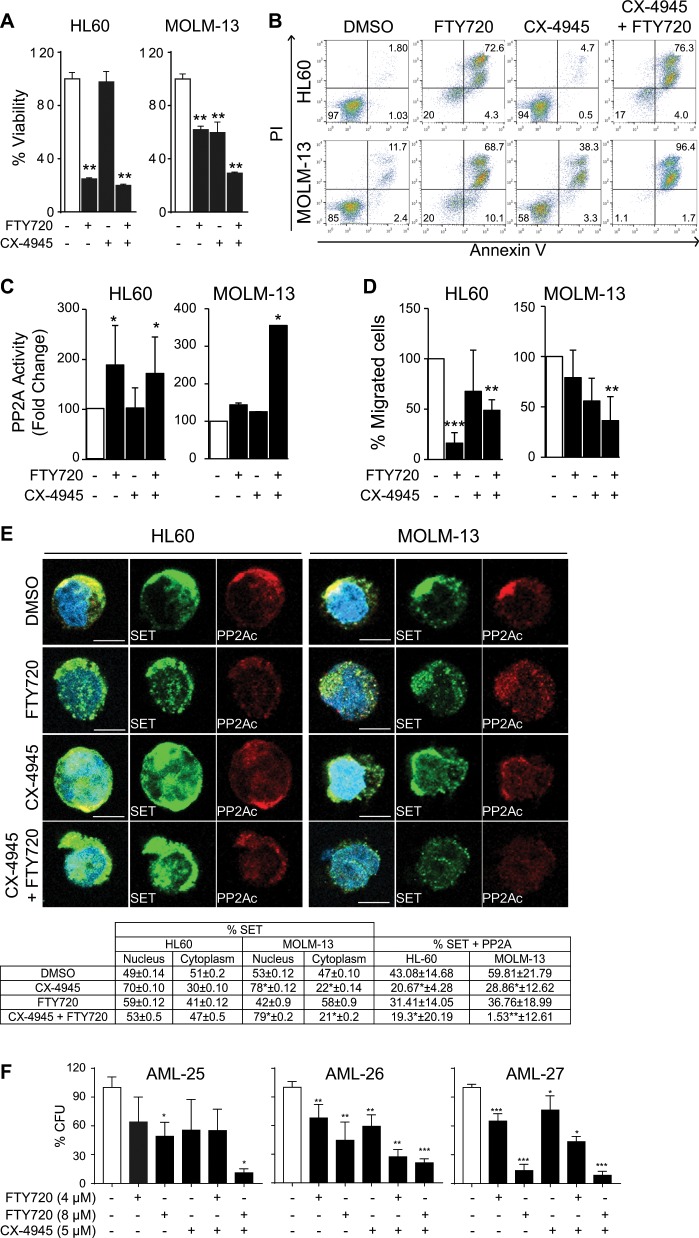
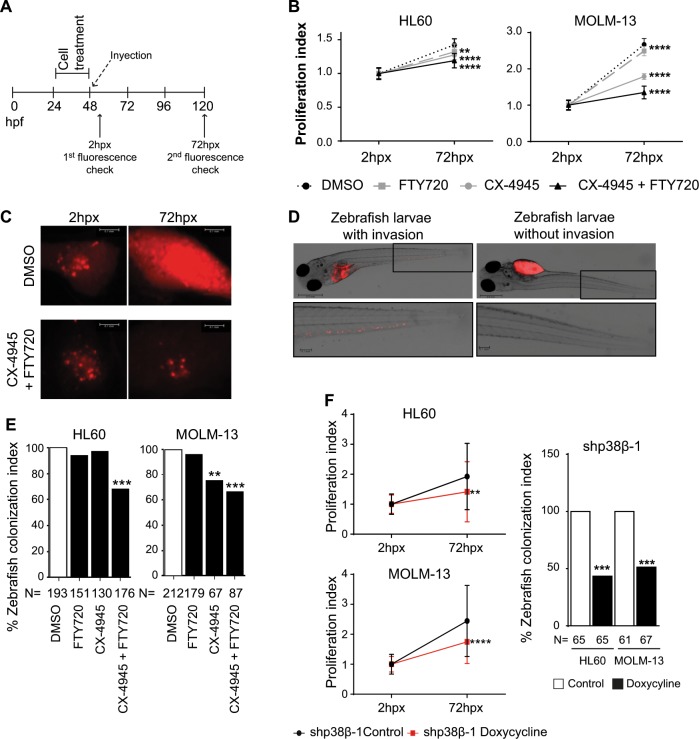
Acute myeloid leukemia (AML) is an aggressive hematologic malignancy. Although novel emerging drugs are available, the overall prognosis remains poor and new therapeutic approaches are required. PP2A phosphatase is a key regulator of cell homeostasis and is recurrently inactivated in AML. The anticancer activity of several PP2A-activating drugs (e.g., FTY720) depends on their interaction with the SET oncoprotein, an endogenous PP2A inhibitor that is overexpressed in 30% of AML cases. Elucidation of SET regulatory mechanisms may therefore provide novel targeted therapies for SET-overexpressing AMLs. Here, we show that upregulation of protein kinase p38β is a common event in AML. We provide evidence that p38β potentiates SET-mediated PP2A inactivation by two mechanisms: facilitating SET cytoplasmic translocation through CK2 phosphorylation, and directly binding to and stabilizing the SET protein. We demonstrate the importance of this new regulatory mechanism in primary AML cells from patients and in zebrafish xenograft models. Accordingly, combination of the CK2 inhibitor CX-4945, which retains SET in the nucleus, and FTY720, which disrupts the SET-PP2A binding in the cytoplasm, significantly reduces the viability and migration of AML cells. In conclusion, we show that the p38β/CK2/SET axis represents a new potential therapeutic pathway in AML patients with SET-dependent PP2A inactivation.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
