

A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose
- PMID: 31914417
- PMCID: PMC6996897
- DOI: 10.1074/jbc.RA119.011471
A synthetic lethal drug combination mimics glucose deprivation-induced cancer cell death in the presence of glucose
Abstract
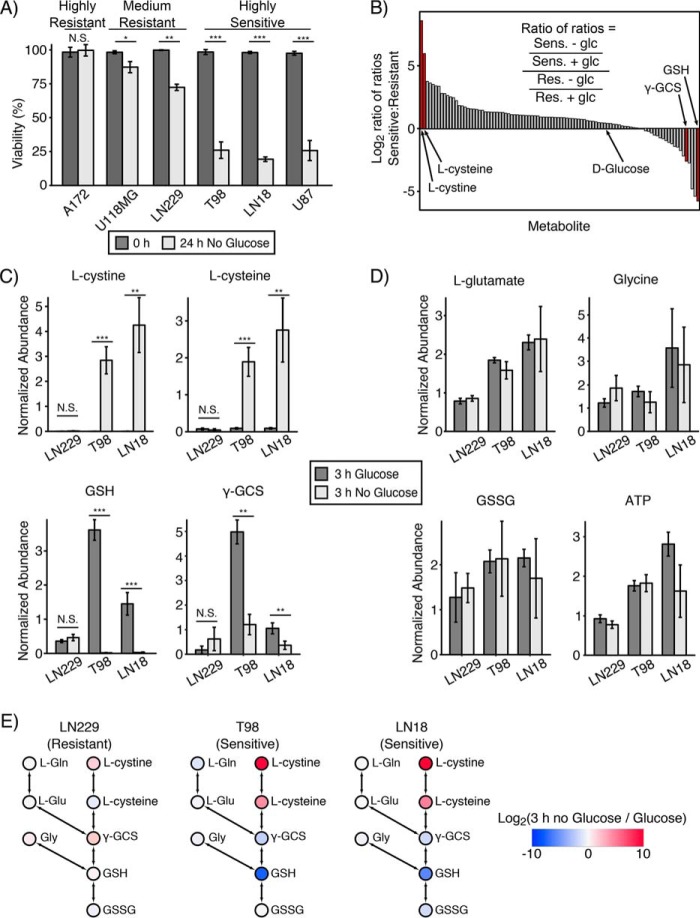
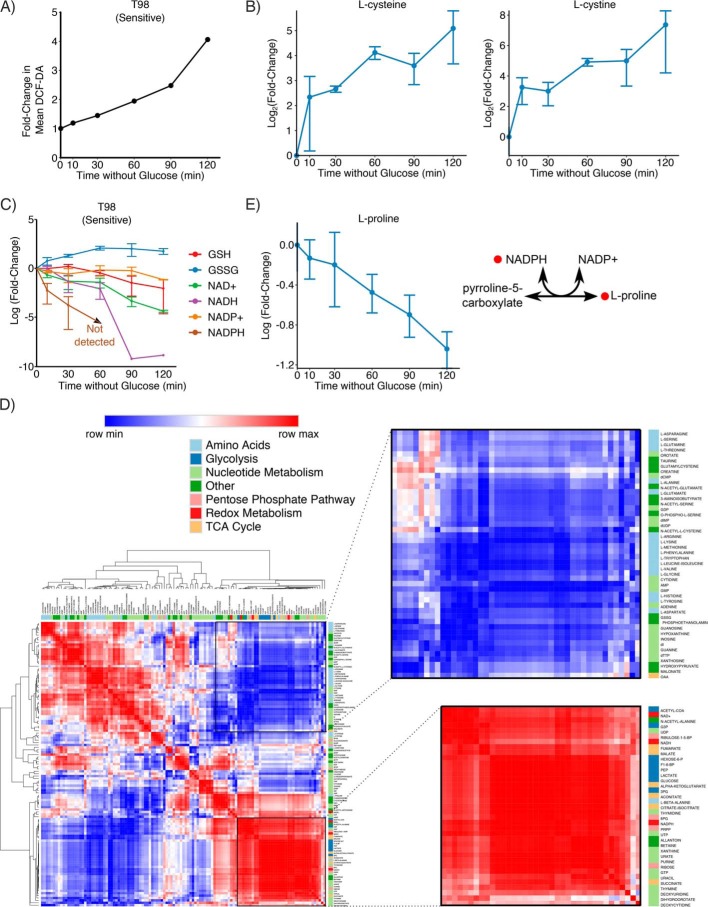
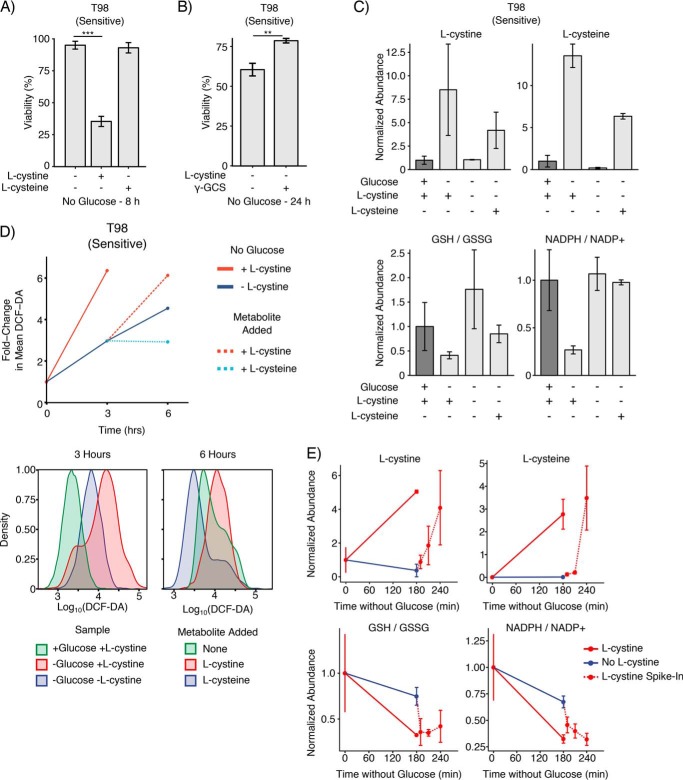
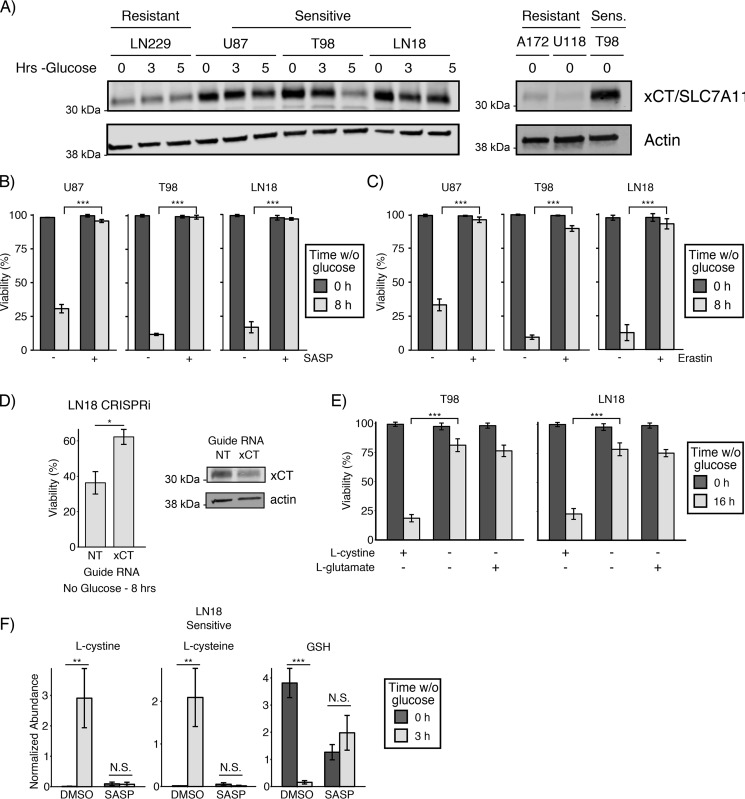
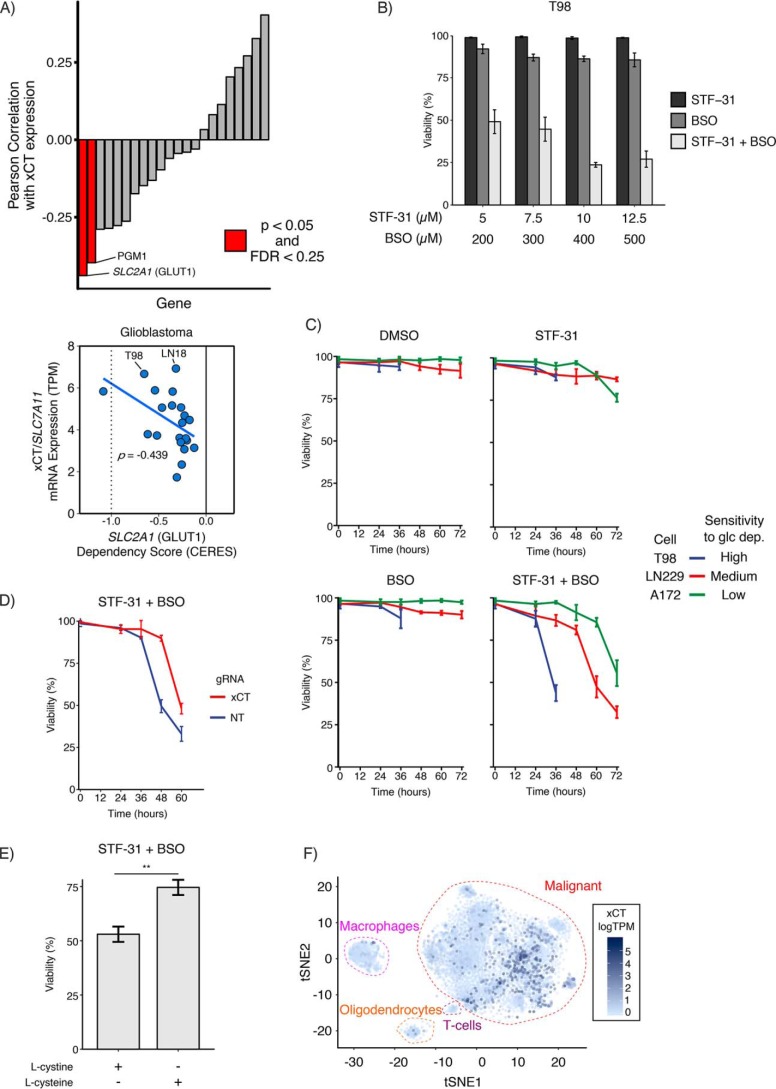
Metabolic reprogramming in cancer cells can increase their dependence on metabolic substrates such as glucose. As such, the vulnerability of cancer cells to glucose deprivation creates an attractive opportunity for therapeutic intervention. Because it is not possible to starve tumors of glucose in vivo, here we sought to identify the mechanisms in glucose deprivation-induced cancer cell death and then designed inhibitor combinations to mimic glucose deprivation-induced cell death. Using metabolomic profiling, we found that cells undergoing glucose deprivation-induced cell death exhibited dramatic accumulation of intracellular l-cysteine and its oxidized dimer, l-cystine, and depletion of the antioxidant GSH. Building on this observation, we show that glucose deprivation-induced cell death is driven not by the lack of glucose, but rather by l-cystine import. Following glucose deprivation, the import of l-cystine and its subsequent reduction to l-cysteine depleted both NADPH and GSH pools, thereby allowing toxic accumulation of reactive oxygen species. Consistent with this model, we found that the glutamate/cystine antiporter (xCT) is required for increased sensitivity to glucose deprivation. We searched for glycolytic enzymes whose expression is essential for the survival of cancer cells with high xCT expression and identified glucose transporter type 1 (GLUT1). Testing a drug combination that co-targeted GLUT1 and GSH synthesis, we found that this combination induces synthetic lethal cell death in high xCT-expressing cell lines susceptible to glucose deprivation. These results indicate that co-targeting GLUT1 and GSH synthesis may offer a potential therapeutic approach for targeting tumors dependent on glucose for survival.
Keywords: L-cystine; NADPH; SLC7A11; Warburg effect; cancer biology; glucose metabolism; metabolomics; redox regulation.
© 2020 Joly et al.
Conflict of interest statement
The authors declare that they have no conflicts of interest with the contents of this article
Figures

References
-
- Cairns R. A., and Mak T. W. (2016) The current state of cancer metabolism. Nat. Rev. Cancer 16, 613–614 10.1038/nrc.2016.100 - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
