

Natural and pathogenic protein sequence variation affecting prion-like domains within and across human proteomes
- PMID: 31914925
- PMCID: PMC6947906
- DOI: 10.1186/s12864-019-6425-3
Natural and pathogenic protein sequence variation affecting prion-like domains within and across human proteomes
Abstract
Background: Impaired proteostatic regulation of proteins with prion-like domains (PrLDs) is associated with a variety of human diseases including neurodegenerative disorders, myopathies, and certain forms of cancer. For many of these disorders, current models suggest a prion-like molecular mechanism of disease, whereby proteins aggregate and spread to neighboring cells in an infectious manner. The development of prion prediction algorithms has facilitated the large-scale identification of PrLDs among "reference" proteomes for various organisms. However, the degree to which intraspecies protein sequence diversity influences predicted prion propensity has not been systematically examined.
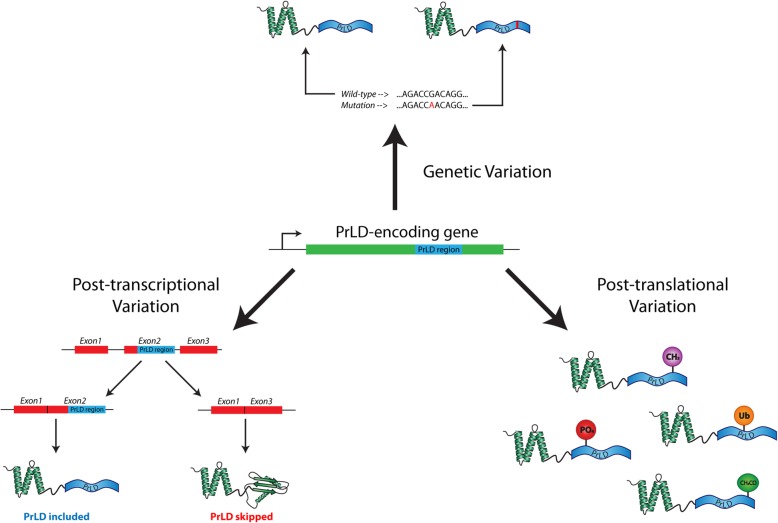
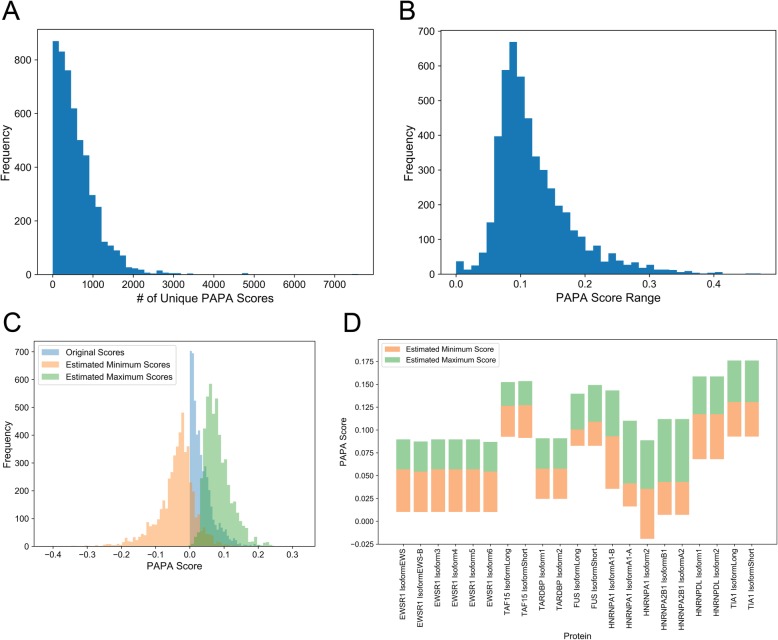
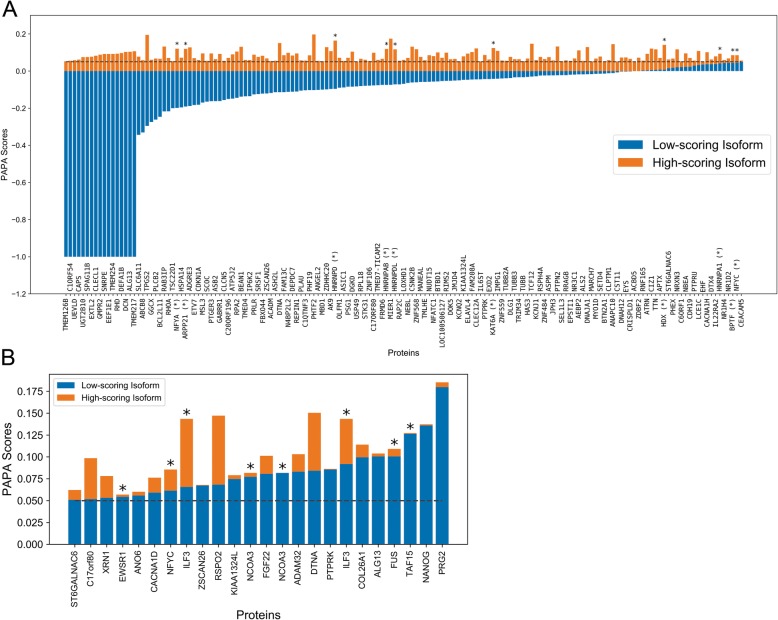
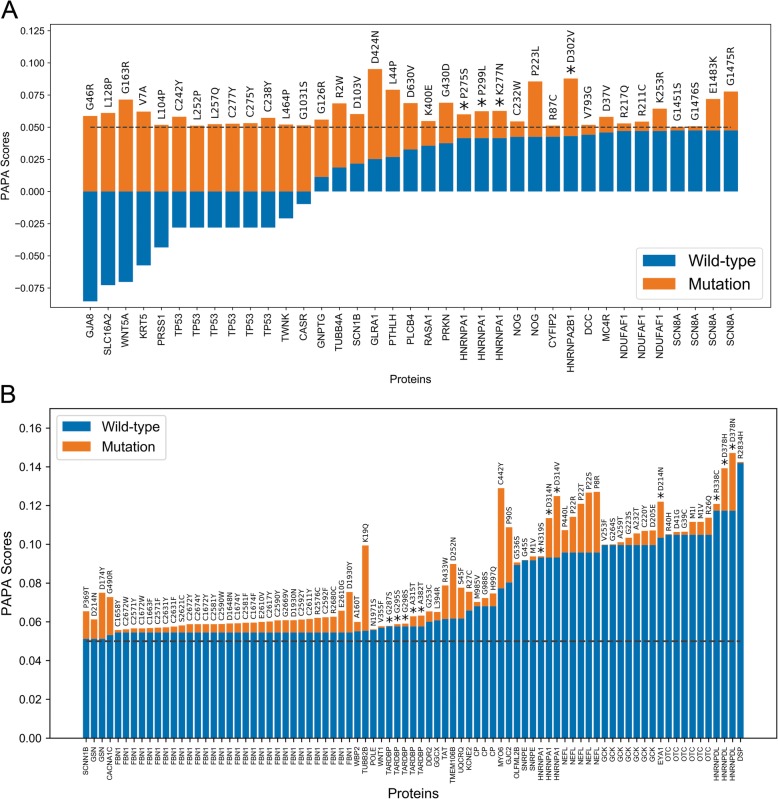
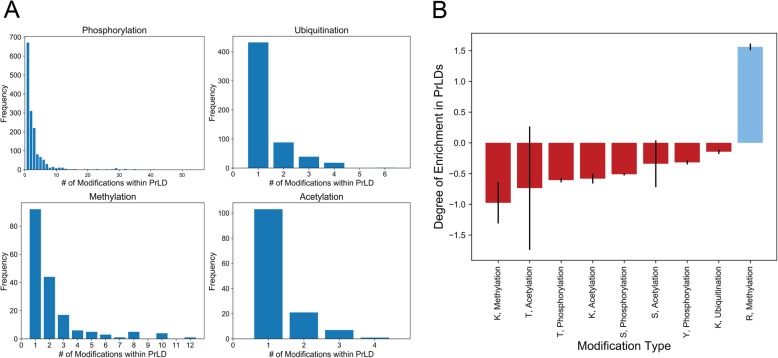
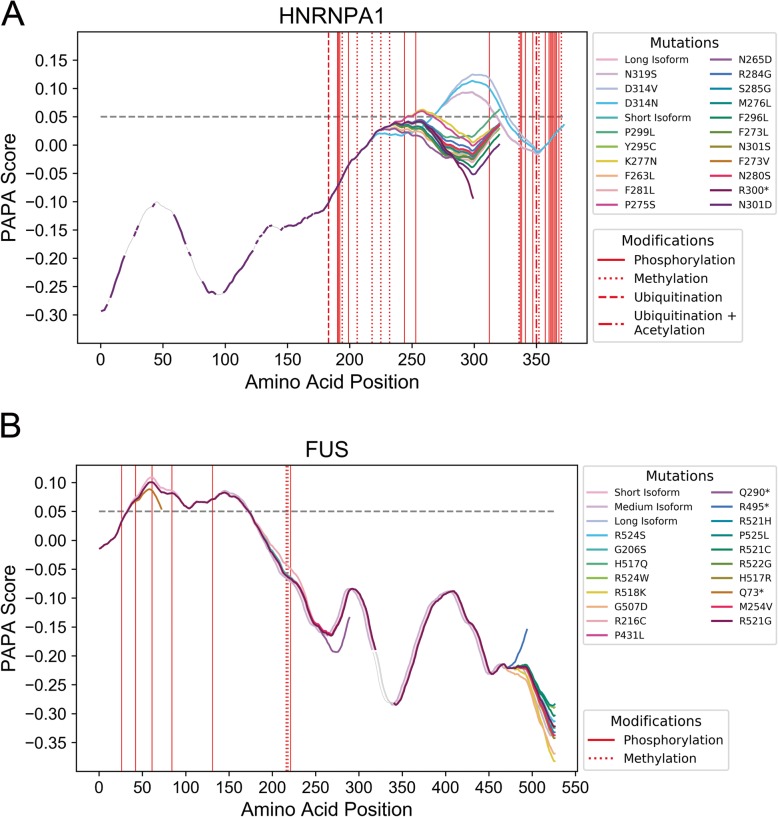
Results: Here, we explore protein sequence variation introduced at genetic, post-transcriptional, and post-translational levels, and its influence on predicted aggregation propensity for human PrLDs. We find that sequence variation is relatively common among PrLDs and in some cases can result in relatively large differences in predicted prion propensity. Sequence variation introduced at the post-transcriptional level (via alternative splicing) also commonly affects predicted aggregation propensity, often by direct inclusion or exclusion of a PrLD. Finally, analysis of a database of sequence variants associated with human disease reveals a number of mutations within PrLDs that are predicted to increase prion propensity.
Conclusions: Our analyses expand the list of candidate human PrLDs, quantitatively estimate the effects of sequence variation on the aggregation propensity of PrLDs, and suggest the involvement of prion-like mechanisms in additional human diseases.
Keywords: Neurodegenerative disease; Prion; Prion prediction; Prion-like domains; Protein aggregation; Sequence variation.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Aggregation and degradation scales for prion-like domains: sequence features and context weigh in.Curr Genet. 2019 Apr;65(2):387-392. doi: 10.1007/s00294-018-0890-0. Epub 2018 Oct 11. Curr Genet. 2019. PMID: 30310993 Review.
-
AMYCO: evaluation of mutational impact on prion-like proteins aggregation propensity.BMC Bioinformatics. 2019 Jan 14;20(1):24. doi: 10.1186/s12859-019-2601-3. BMC Bioinformatics. 2019. PMID: 30642249 Free PMC article.
-
Perfecting prediction of mutational impact on the aggregation propensity of the ALS-associated hnRNPA2 prion-like protein.FEBS Lett. 2017 Jul;591(13):1966-1971. doi: 10.1002/1873-3468.12698. Epub 2017 Jun 18. FEBS Lett. 2017. PMID: 28542905
-
Prion-like domains as epigenetic regulators, scaffolds for subcellular organization, and drivers of neurodegenerative disease.Brain Res. 2016 Sep 15;1647:9-18. doi: 10.1016/j.brainres.2016.02.037. Epub 2016 Mar 18. Brain Res. 2016. PMID: 26996412 Free PMC article. Review.
-
Composition-based prediction and rational manipulation of prion-like domain recruitment to stress granules.Proc Natl Acad Sci U S A. 2020 Mar 17;117(11):5826-5835. doi: 10.1073/pnas.1912723117. Epub 2020 Mar 3. Proc Natl Acad Sci U S A. 2020. PMID: 32127480 Free PMC article.
Cited by
-
Prion-like proteins: from computational approaches to proteome-wide analysis.FEBS Open Bio. 2021 Sep;11(9):2400-2417. doi: 10.1002/2211-5463.13213. Epub 2021 Jun 17. FEBS Open Bio. 2021. PMID: 34057308 Free PMC article. Review.
-
Saccharomyces cerevisiae in neuroscience: how unicellular organism helps to better understand prion protein?Neural Regen Res. 2021 Mar;16(3):489-495. doi: 10.4103/1673-5374.293137. Neural Regen Res. 2021. PMID: 32985470 Free PMC article. Review.
-
The Quest for Cellular Prion Protein Functions in the Aged and Neurodegenerating Brain.Cells. 2020 Mar 2;9(3):591. doi: 10.3390/cells9030591. Cells. 2020. PMID: 32131451 Free PMC article. Review.
-
Generalizable Compositional Features Influencing the Proteostatic Fates of Polar Low-Complexity Domains.Int J Mol Sci. 2021 Aug 19;22(16):8944. doi: 10.3390/ijms22168944. Int J Mol Sci. 2021. PMID: 34445649 Free PMC article.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
