

Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma
- PMID: 31915197
- PMCID: PMC7275250
- DOI: 10.1158/2159-8290.CD-19-0116
Selective Inhibition of HDAC3 Targets Synthetic Vulnerabilities and Activates Immune Surveillance in Lymphoma
Abstract
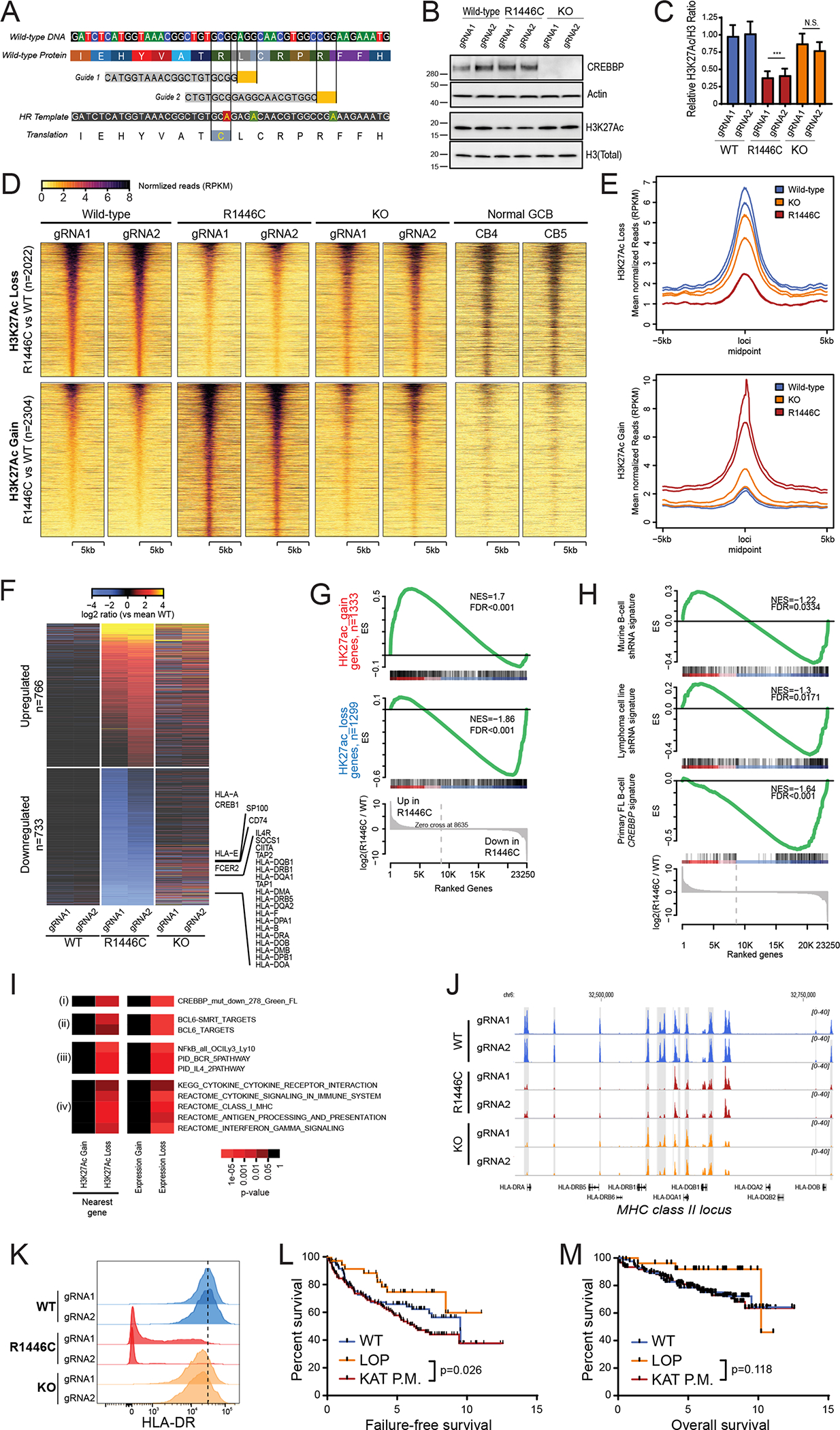
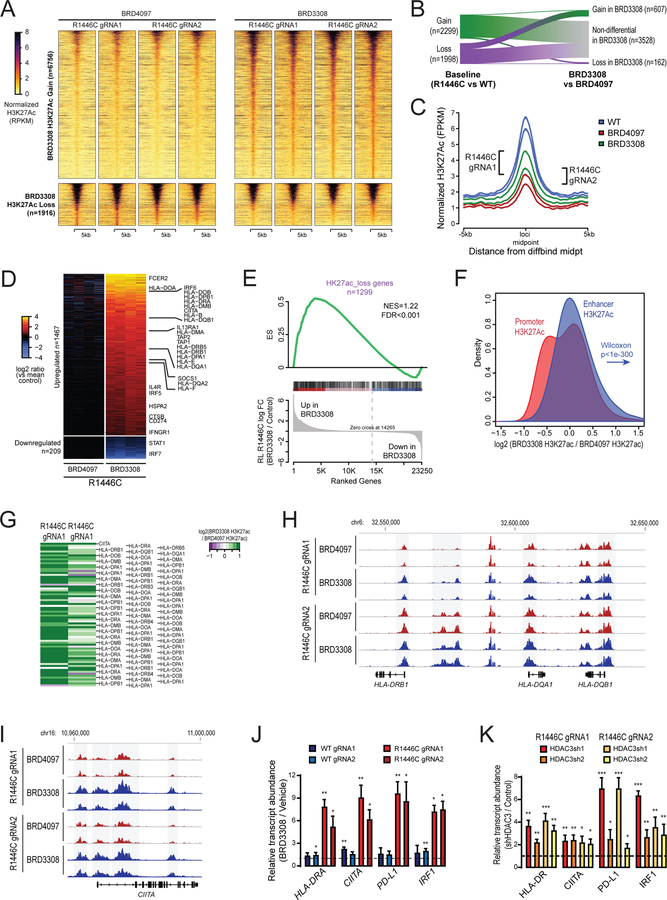
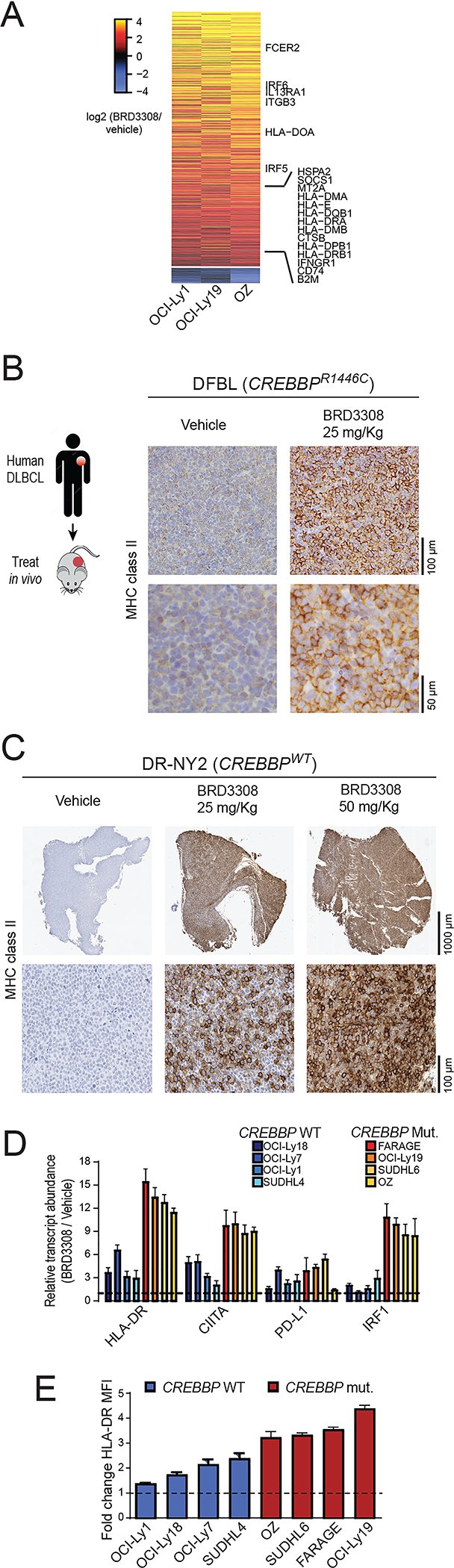
CREBBP mutations are highly recurrent in B-cell lymphomas and either inactivate its histone acetyltransferase (HAT) domain or truncate the protein. Herein, we show that these two classes of mutations yield different degrees of disruption of the epigenome, with HAT mutations being more severe and associated with inferior clinical outcome. Genes perturbed by CREBBP mutation are direct targets of the BCL6-HDAC3 onco-repressor complex. Accordingly, we show that HDAC3-selective inhibitors reverse CREBBP-mutant aberrant epigenetic programming, resulting in: (i) growth inhibition of lymphoma cells through induction of BCL6 target genes such as CDKN1A and (ii) restoration of immune surveillance due to induction of BCL6-repressed IFN pathway and antigen-presenting genes. By reactivating these genes, exposure to HDAC3 inhibitors restored the ability of tumor-infiltrating lymphocytes to kill DLBCL cells in an MHC class I and II-dependent manner, and synergized with PD-L1 blockade in a syngeneic model in vivo. Hence, HDAC3 inhibition represents a novel mechanism-based immune epigenetic therapy for CREBBP-mutant lymphomas. SIGNIFICANCE: We have leveraged the molecular characterization of different types of CREBBP mutations to define a rational approach for targeting these mutations through selective inhibition of HDAC3. This represents an attractive therapeutic avenue for targeting synthetic vulnerabilities in CREBBP-mutant cells in tandem with promoting antitumor immunity.This article is highlighted in the In This Issue feature, p. 327.
©2020 American Association for Cancer Research.
Conflict of interest statement
Figures




References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
