

Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition
- PMID: 31915379
- PMCID: PMC7308074
- DOI: 10.1038/s41586-019-1884-x
Rapid non-uniform adaptation to conformation-specific KRAS(G12C) inhibition
Abstract
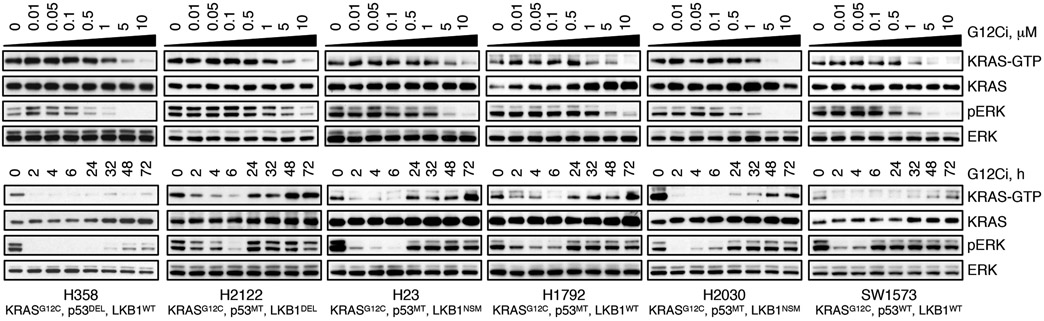
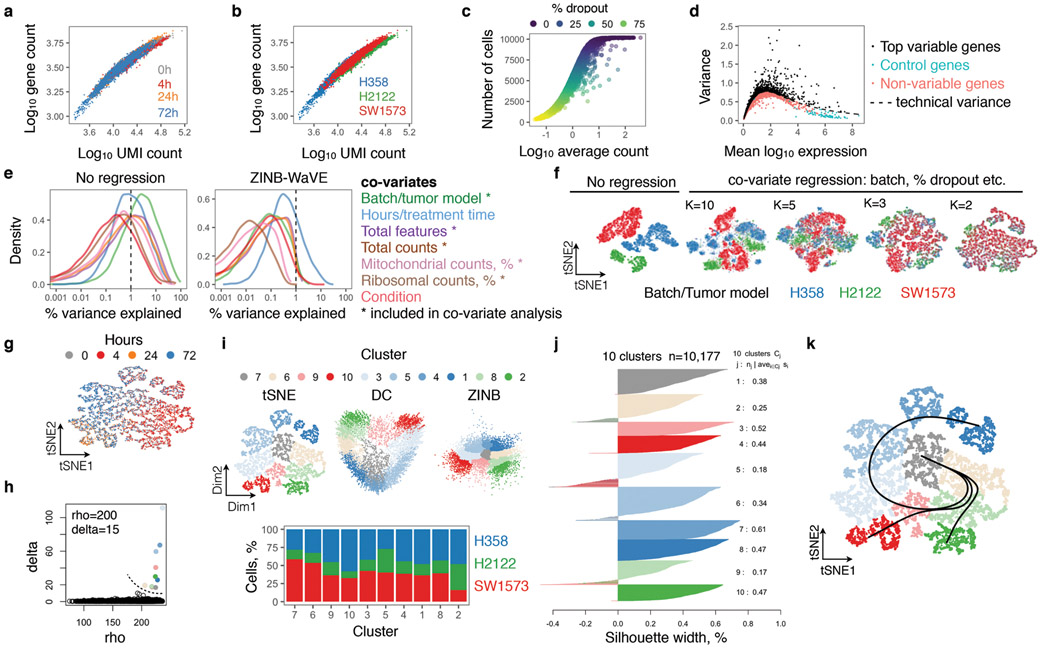
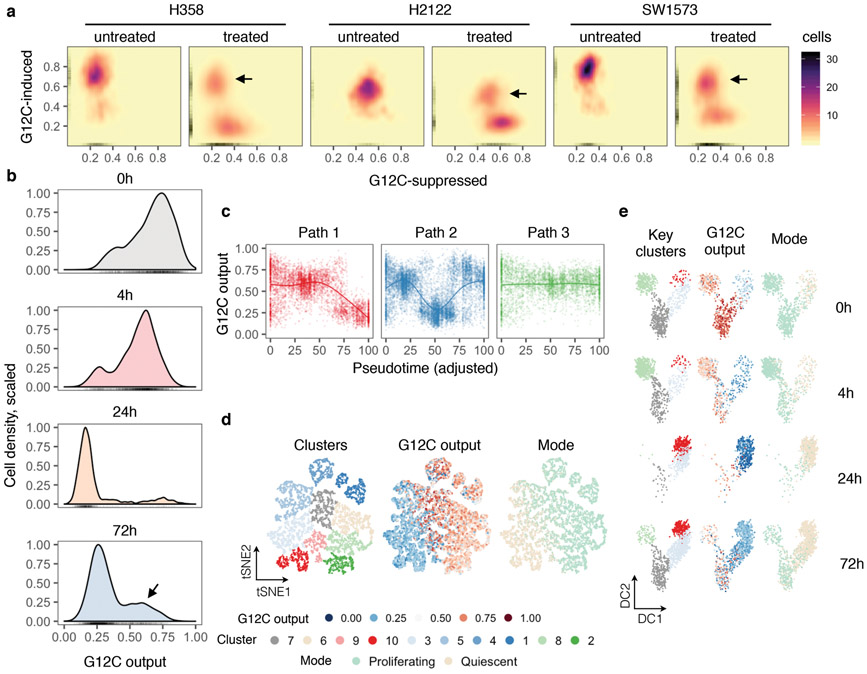
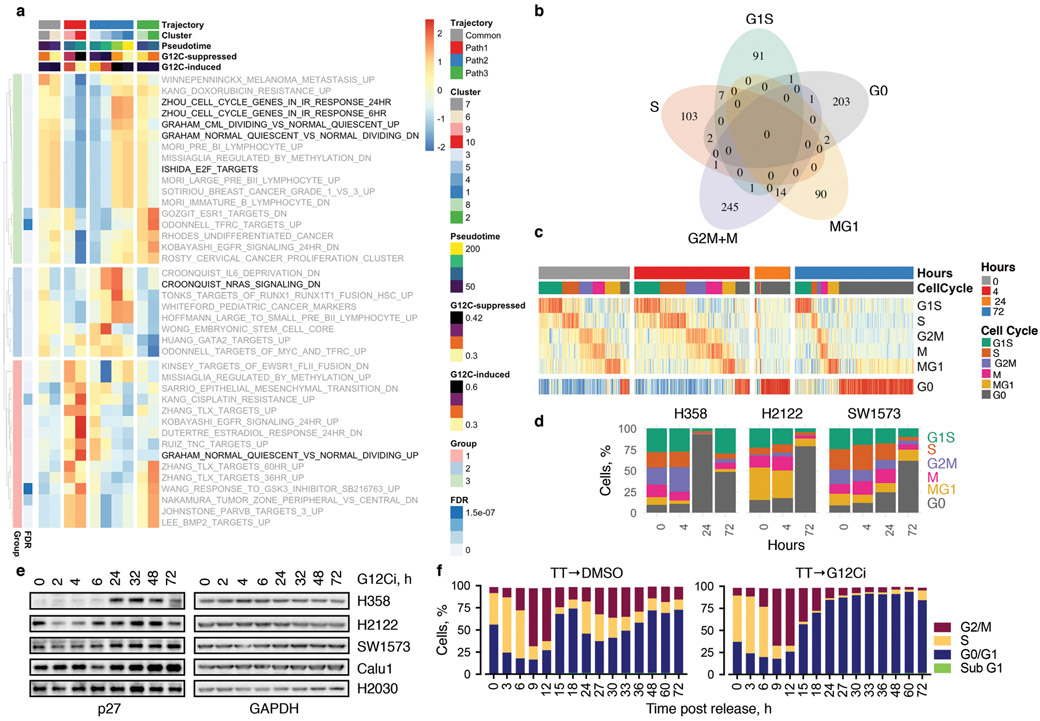
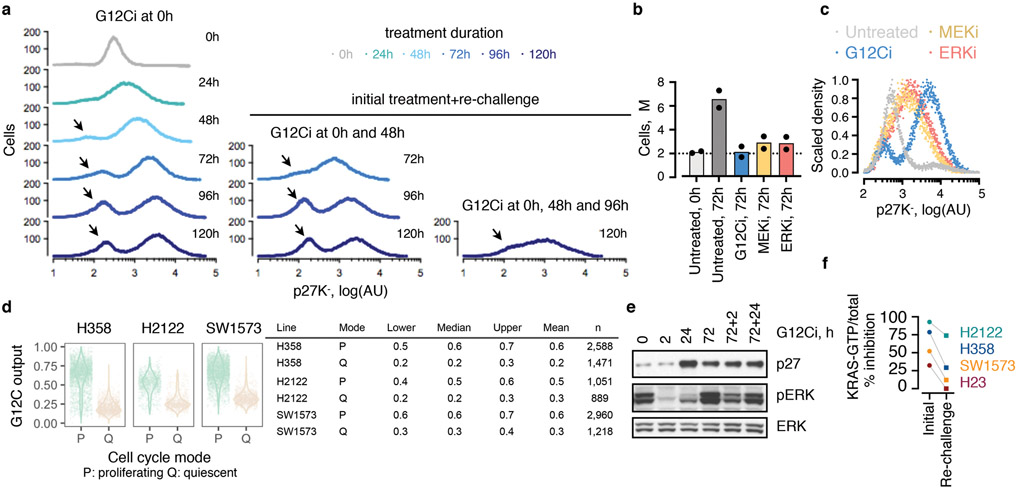
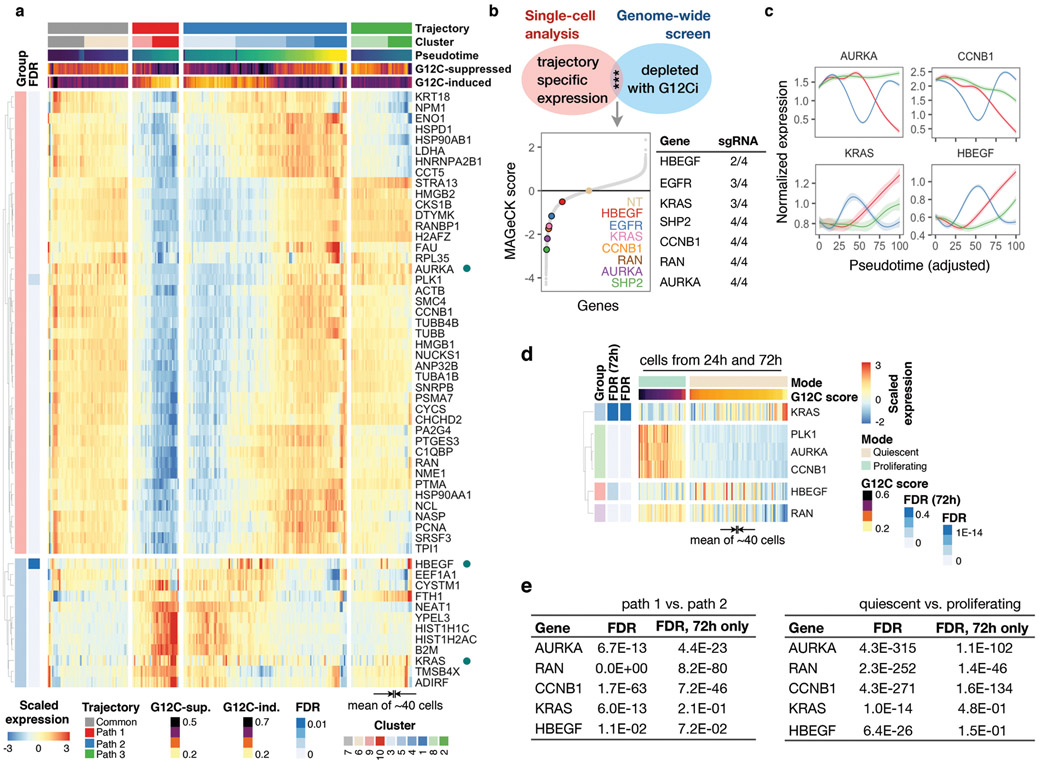
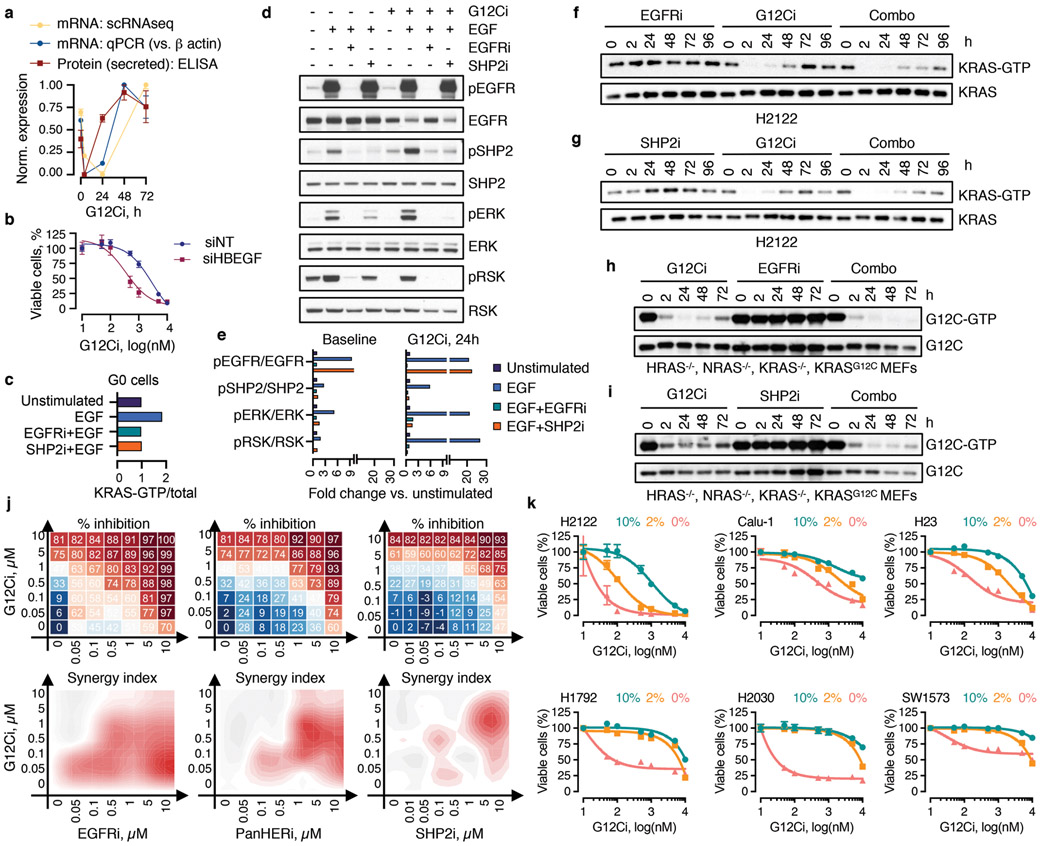
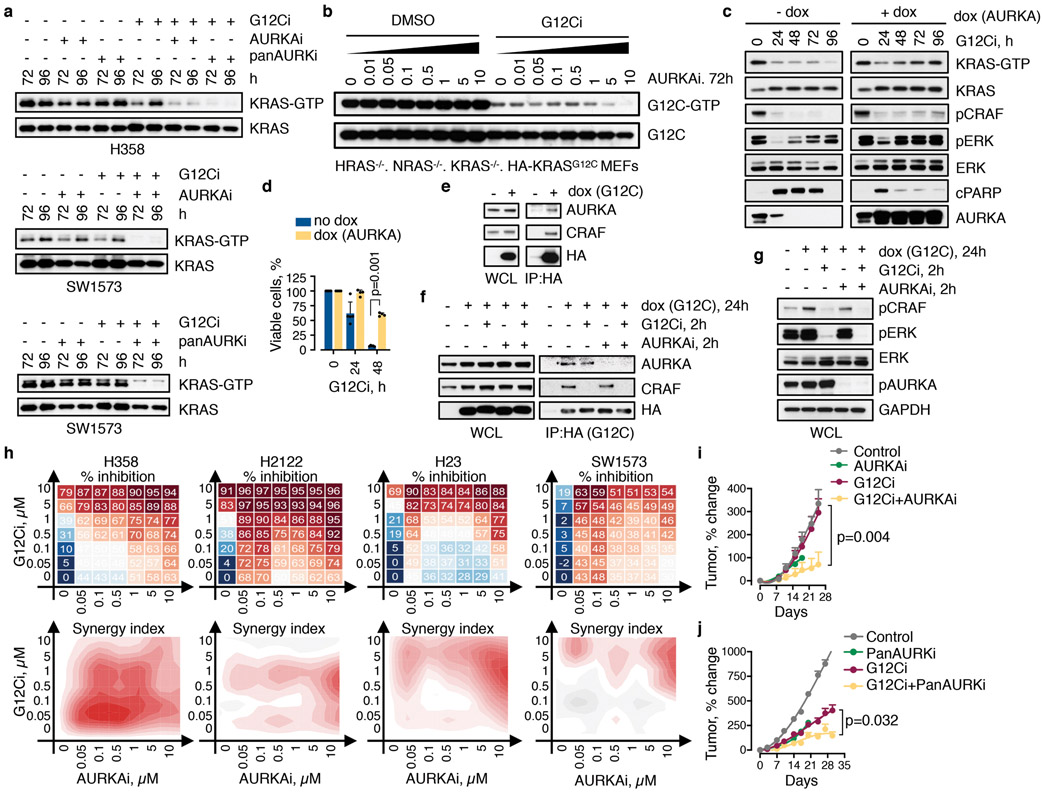
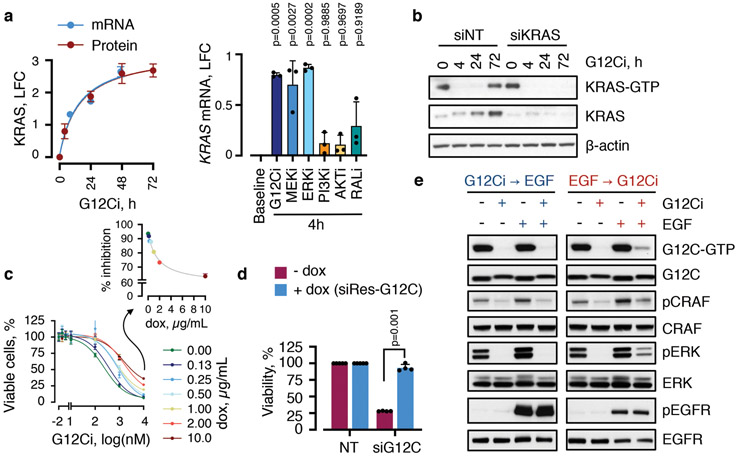
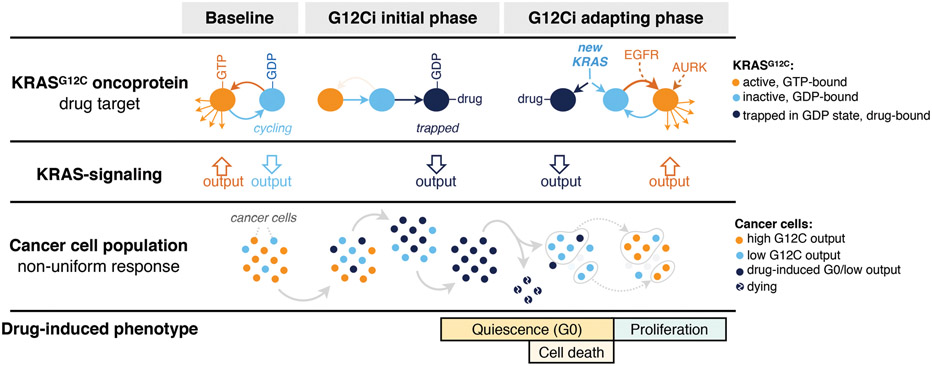
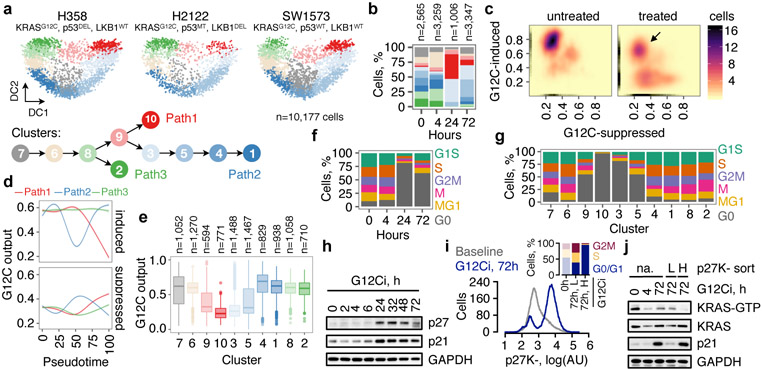
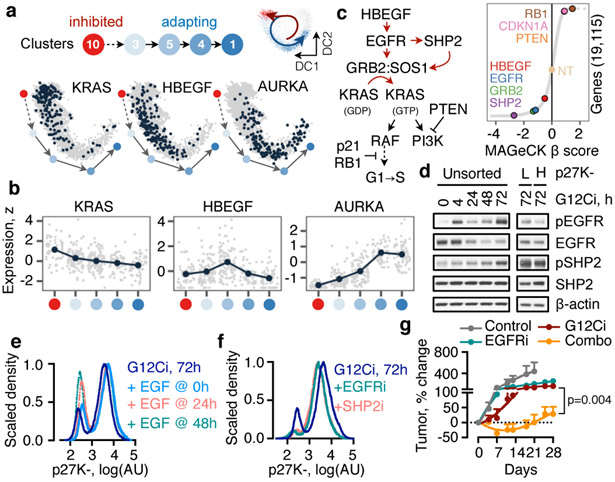
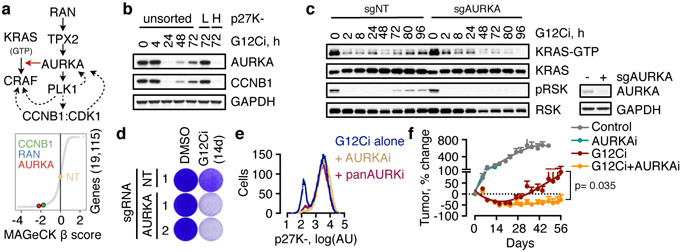
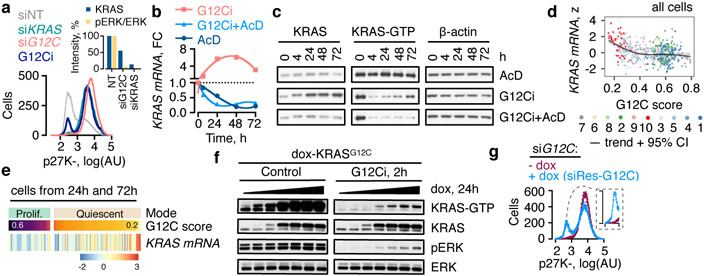
KRAS GTPases are activated in one-third of cancers, and KRAS(G12C) is one of the most common activating alterations in lung adenocarcinoma1,2. KRAS(G12C) inhibitors3,4 are in phase-I clinical trials and early data show partial responses in nearly half of patients with lung cancer. How cancer cells bypass inhibition to prevent maximal response to therapy is not understood. Because KRAS(G12C) cycles between an active and inactive conformation4-6, and the inhibitors bind only to the latter, we tested whether isogenic cell populations respond in a non-uniform manner by studying the effect of treatment at a single-cell resolution. Here we report that, shortly after treatment, some cancer cells are sequestered in a quiescent state with low KRAS activity, whereas others bypass this effect to resume proliferation. This rapid divergent response occurs because some quiescent cells produce new KRAS(G12C) in response to suppressed mitogen-activated protein kinase output. New KRAS(G12C) is maintained in its active, drug-insensitive state by epidermal growth factor receptor and aurora kinase signalling. Cells without these adaptive changes-or cells in which these changes are pharmacologically inhibited-remain sensitive to drug treatment, because new KRAS(G12C) is either not available or exists in its inactive, drug-sensitive state. The direct targeting of KRAS oncoproteins has been a longstanding objective in precision oncology. Our study uncovers a flexible non-uniform fitness mechanism that enables groups of cells within a population to rapidly bypass the effect of treatment. This adaptive process must be overcome if we are to achieve complete and durable responses in the clinic.
Figures
Comment in
-
Newly Synthesized KRASG12C Mediates Escape from KRASG12C Inhibition.Cancer Discov. 2020 Mar;10(3):341. doi: 10.1158/2159-8290.CD-RW2020-010. Epub 2020 Jan 17. Cancer Discov. 2020. PMID: 31953242
-
Resistance looms for KRASG12C inhibitors.Nat Med. 2020 Feb;26(2):169-170. doi: 10.1038/s41591-020-0765-z. Nat Med. 2020. PMID: 32020086 No abstract available.
References
-
- Patricelli MP et al. Selective Inhibition of Oncogenic KRAS Output with Small Molecules Targeting the Inactive State. Cancer discovery 6, 316–329, doi: 10.1158/2159-8290.CD-15-1105 (2016). - DOI - PubMed
Additional full text HTML references:
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
