

Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein
- PMID: 31920150
- PMCID: PMC7595620
- DOI: 10.1080/15548627.2020.1714209
Autophagy-dependent ferroptosis drives tumor-associated macrophage polarization via release and uptake of oncogenic KRAS protein
Abstract
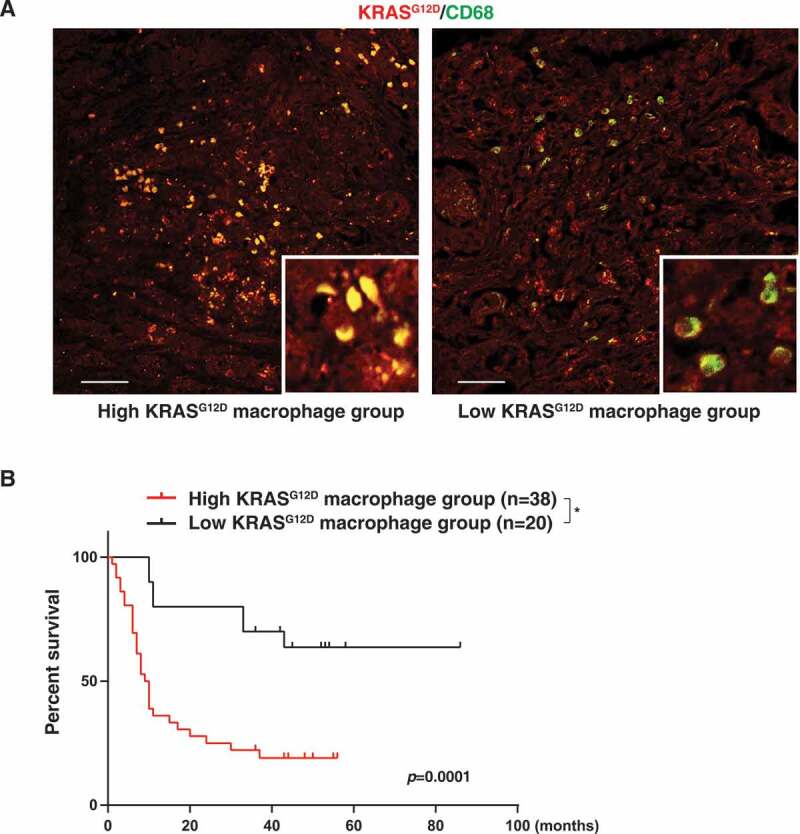
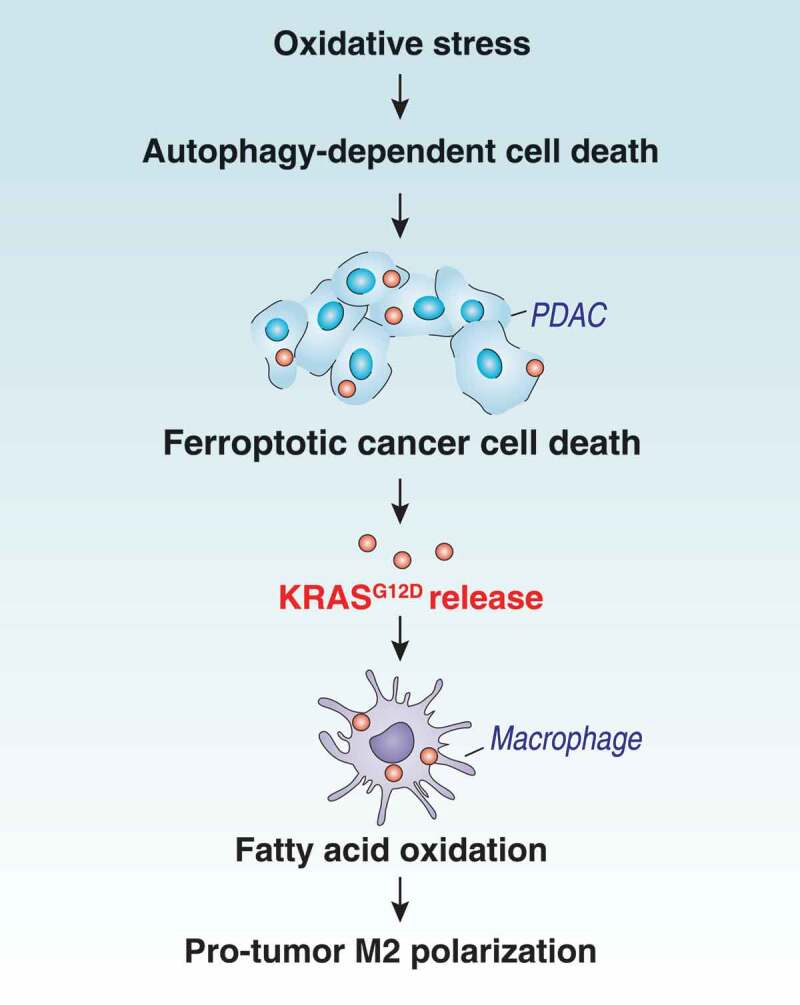
KRAS is the most frequently mutated oncogene in human neoplasia. Despite a large investment to understand the effects of KRAS mutation in cancer cells, the direct effects of the oncogenetic KRAS activation on immune cells remain elusive. Here, we report that extracellular KRASG12D is essential for pancreatic tumor-associated macrophage polarization. Oxidative stress induces KRASG12D protein release from cancer cells succumbing to autophagy-dependent ferroptosis. Extracellular KRASG12D packaged into exosomes then is taken up by macrophages through an AGER-dependent mechanism. KRASG12D causes macrophages to switch to an M2-like pro-tumor phenotype via STAT3-dependent fatty acid oxidation. Consequently, the disruption of KRASG12D release and uptake can abolish the macrophage-mediated stimulation of pancreatic adenocarcinomas in mouse models. Importantly, the level of KRASG12D expression in macrophages correlates with poor survival in pancreatic cancer patients. These findings not only identify extracellular KRASG12D as a key mediator of cancer cell-macrophage communication, but also provide a novel KRAS-targeted anticancer strategy. Abbreviations: DAMP, damage-associated molecular pattern; PBMCMs, peripheral blood mononuclear cell-derived macrophages; PDAC, pancreatic ductal adenocarcinoma; s.c., subcutaneously; TAMs, tumor-associated macrophages; TME, tumor microenvironment.
Keywords: DAMP; KRAS; autophagy; exosomes; ferroptosis; macrophage.
Conflict of interest statement
No potential conflict of interest was reported by the authors.
Figures
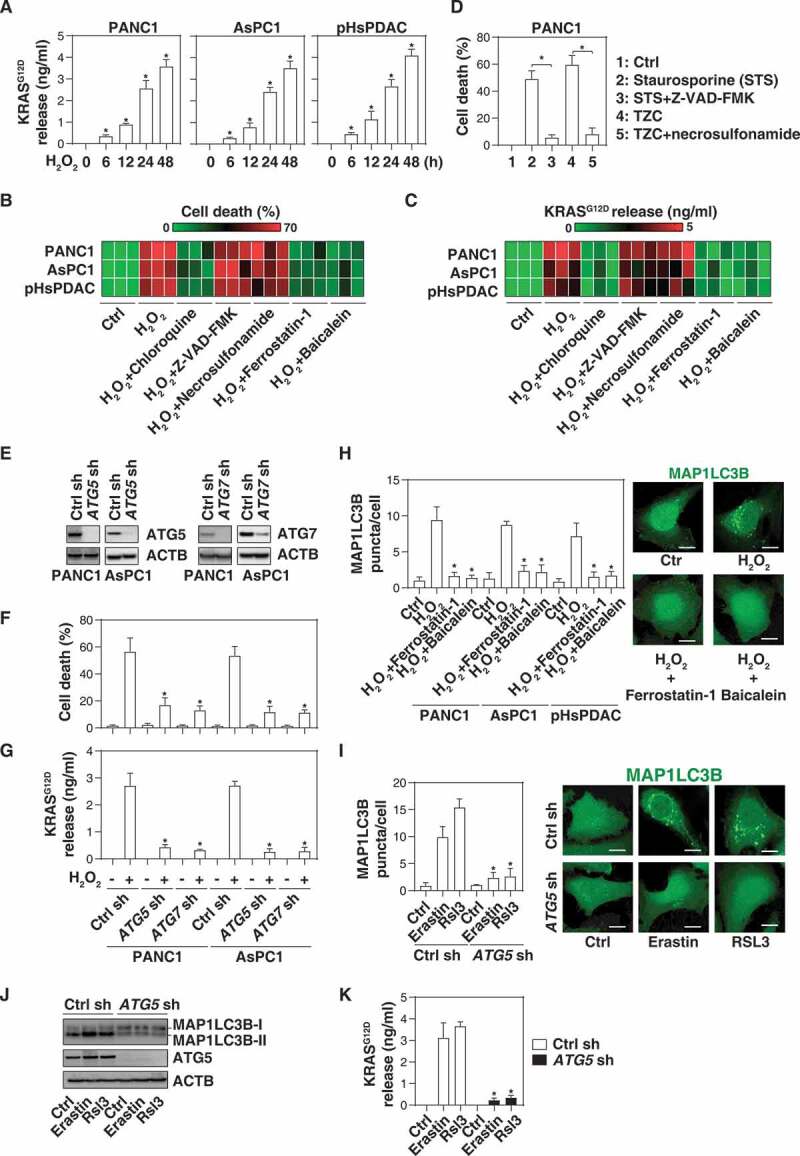
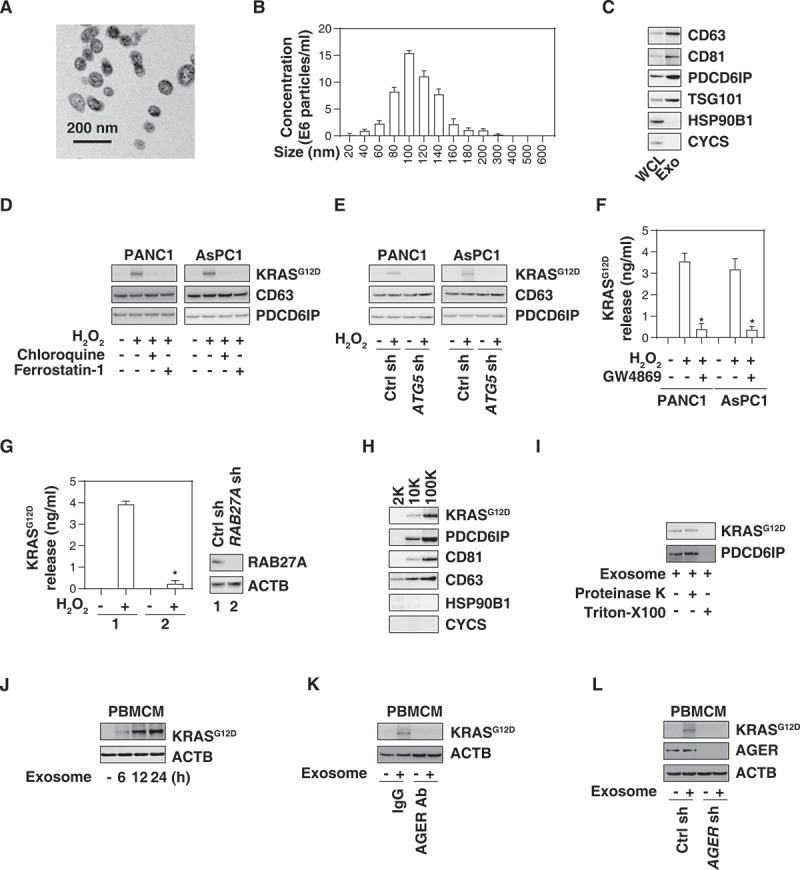
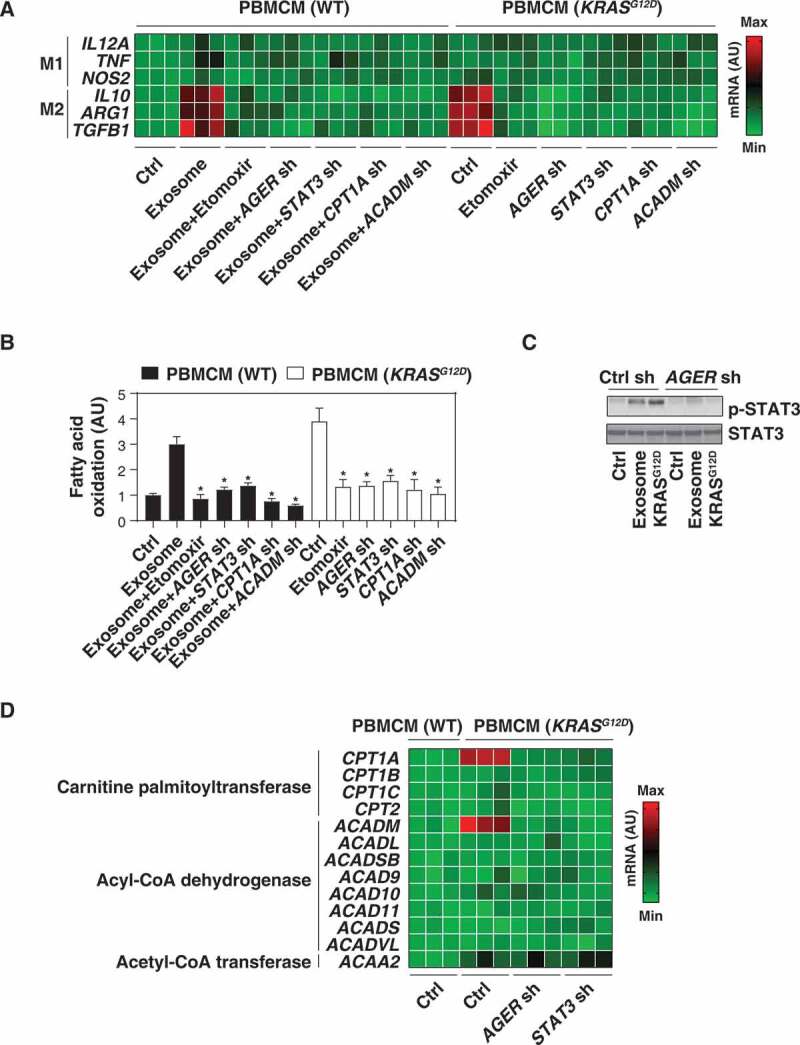
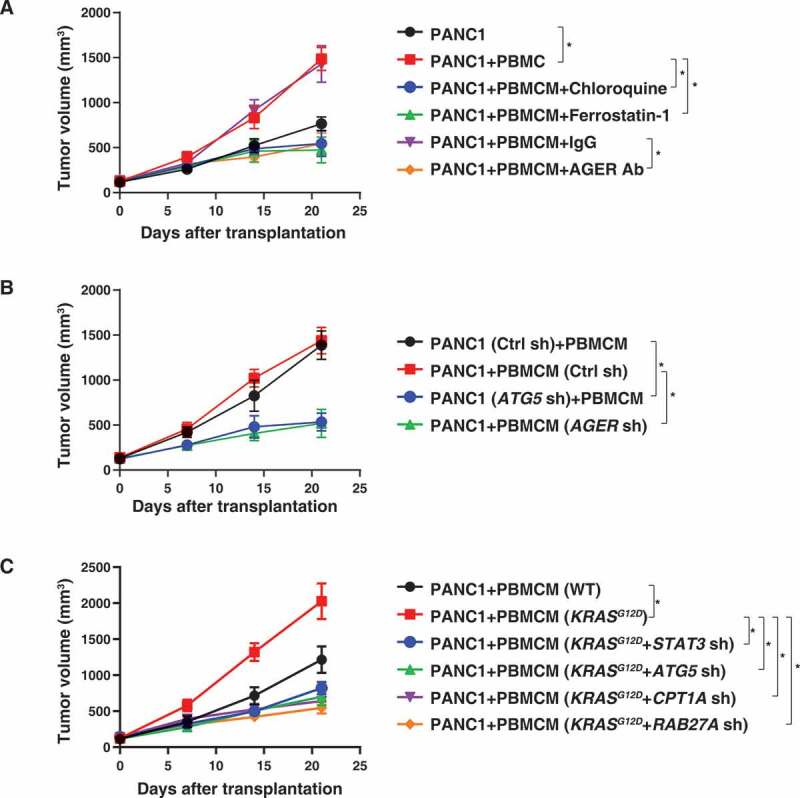
References
-
- Wellenstein MD, de Visser KE.. Cancer-cell-intrinsic mechanisms shaping the tumor immune landscape. Immunity. 2018;48:399–416. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous