

Co-expression Networks Identify DHX15 RNA Helicase as a B Cell Regulatory Factor
- PMID: 31921164
- PMCID: PMC6915936
- DOI: 10.3389/fimmu.2019.02903
Co-expression Networks Identify DHX15 RNA Helicase as a B Cell Regulatory Factor
Abstract
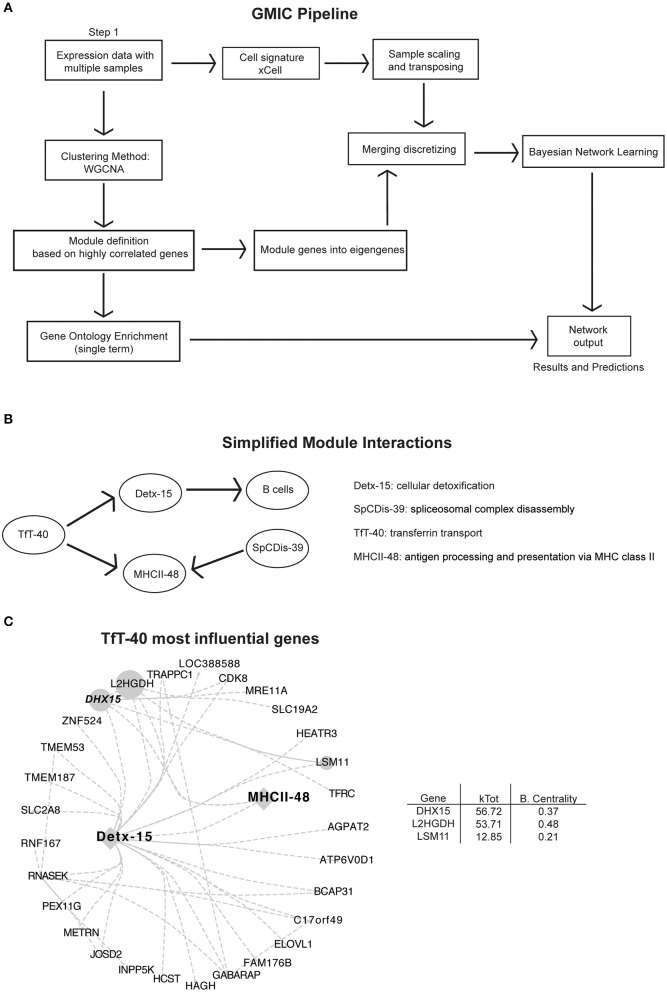
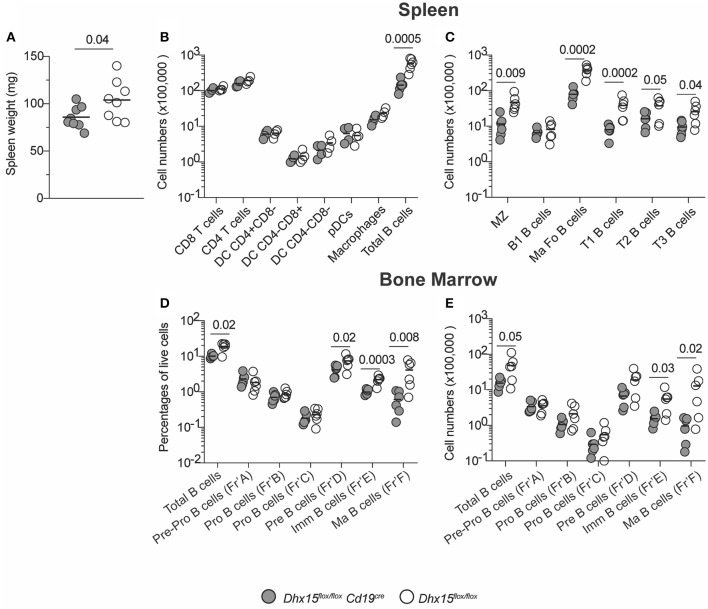
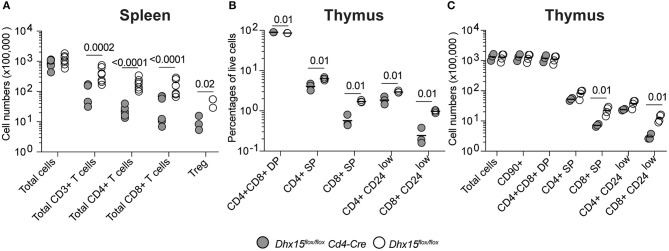
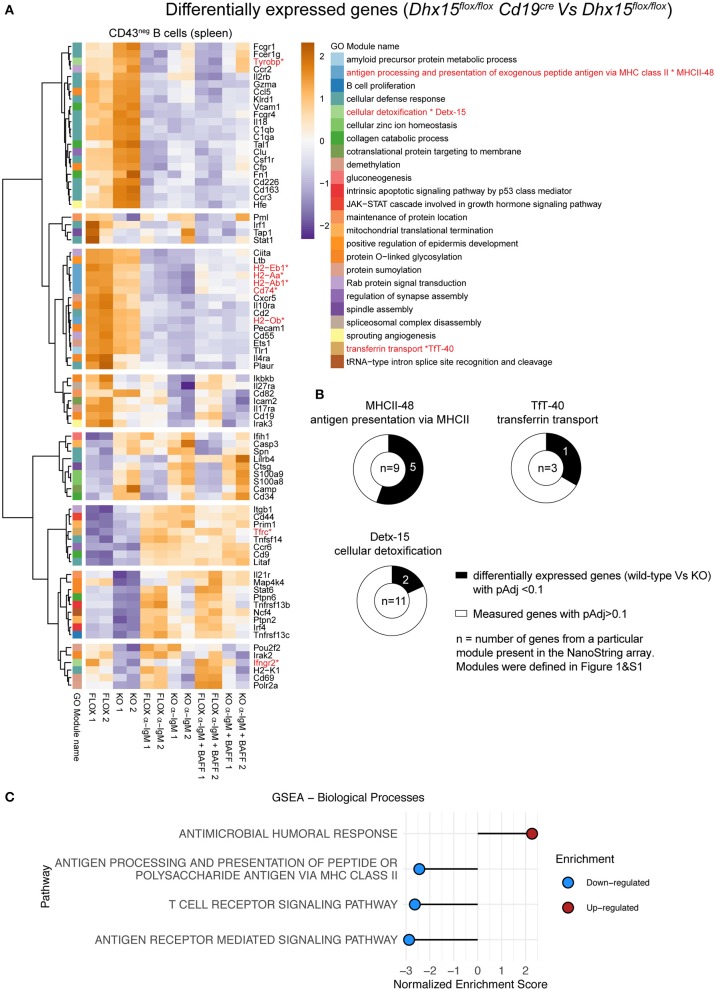
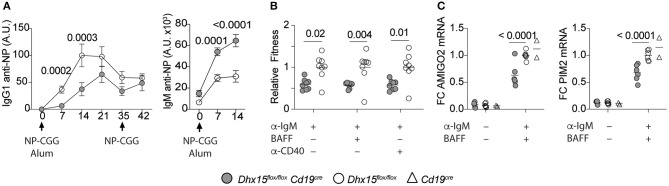
Genome-wide co-expression analysis is often used for annotating novel gene functions from high-dimensional data. Here, we developed an R package with a Shiny visualization app that creates immuno-networks from RNAseq data using a combination of Weighted Gene Co-expression Network Analysis (WGCNA), xCell immune cell signatures, and Bayesian Network Learning. Using a large publicly available RNAseq dataset we generated a Gene Module-Immune Cell (GMIC) network that predicted causal relationships between DEAH-box RNA helicase (DHX)15 and genes associated with humoral immunity, suggesting that DHX15 may regulate B cell fate. Deletion of DHX15 in mouse B cells led to impaired lymphocyte development, reduced peripheral B cell numbers, and dysregulated expression of genes linked to antibody-mediated immune responses similar to the genes predicted by the GMIC network. Moreover, antigen immunization of mice demonstrated that optimal primary IgG1 responses required DHX15. Intrinsic expression of DHX15 was necessary for proliferation and survival of activated of B cells. Altogether, these results support the use of co-expression networks to elucidate fundamental biological processes.
Keywords: B cell; Bayesian network; DHX15; RNA helicase; WGCNA; antibodies.
Copyright © 2019 Detanico, Virgen-Slane, Steen-Fuentes, Lin, Rhode-Kurnow, Chappell, Correa, DiCandido, Mbow, Li and Ware.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases