

Benchmark and Parameter Sensitivity Analysis of Single-Cell RNA Sequencing Clustering Methods
- PMID: 31921297
- PMCID: PMC6918801
- DOI: 10.3389/fgene.2019.01253
Benchmark and Parameter Sensitivity Analysis of Single-Cell RNA Sequencing Clustering Methods
Abstract
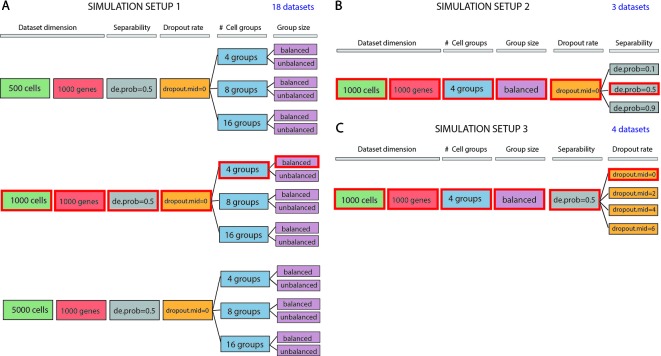
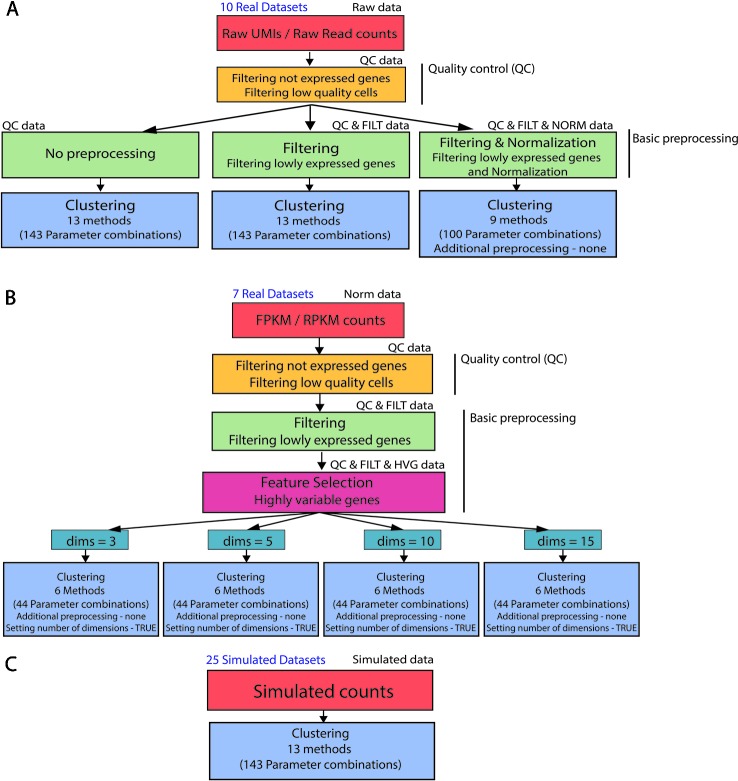
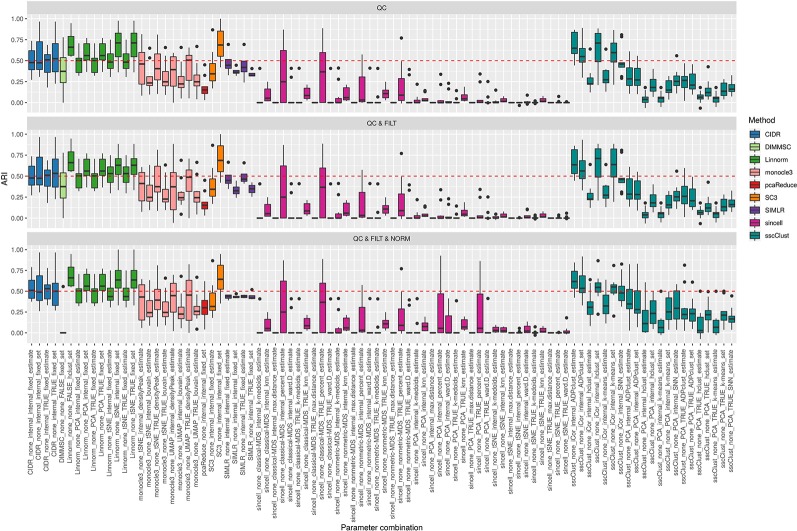
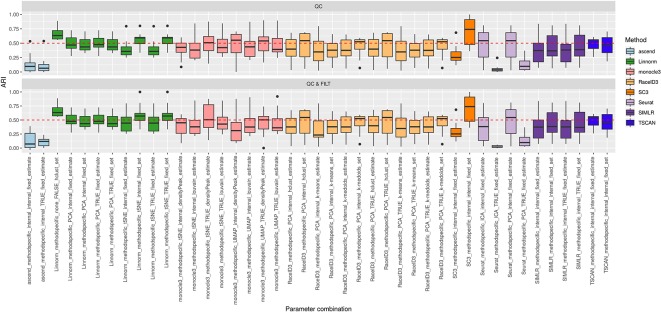
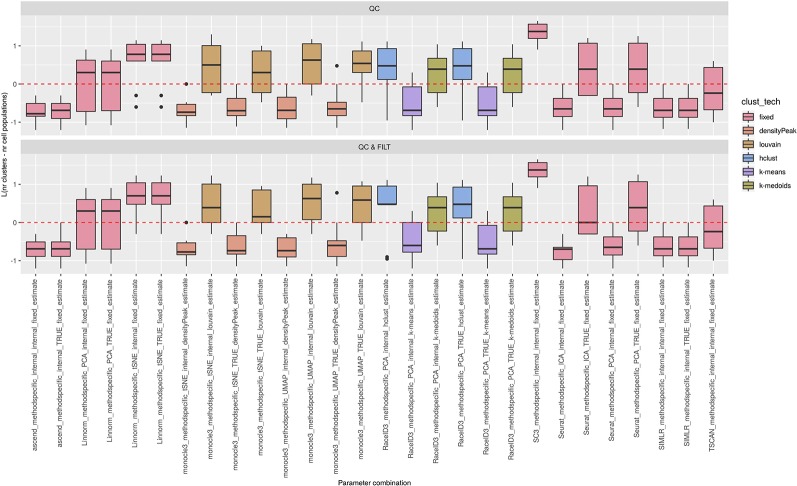
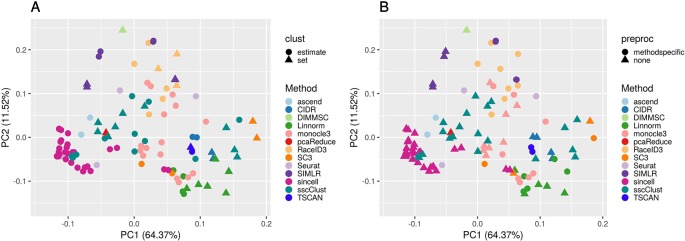
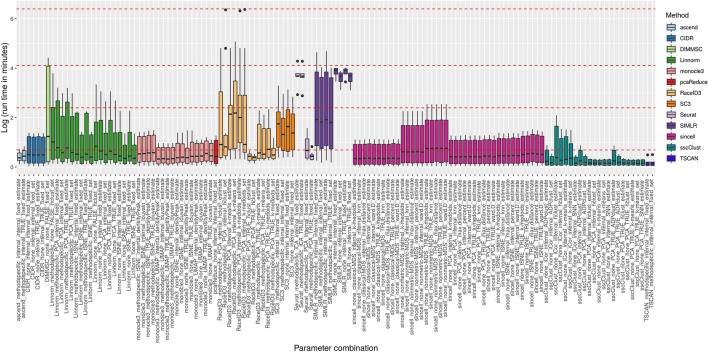
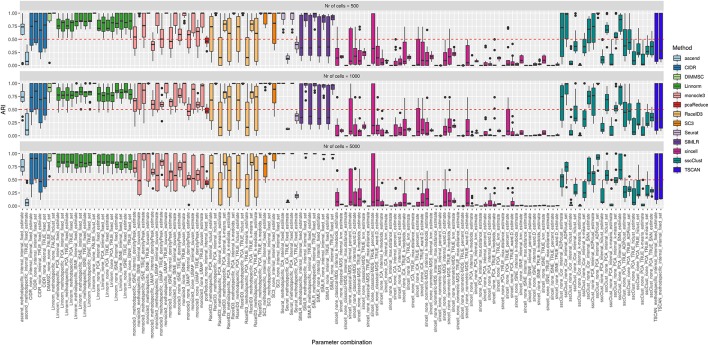
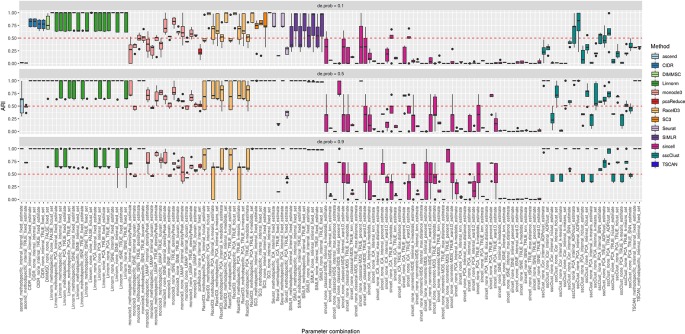
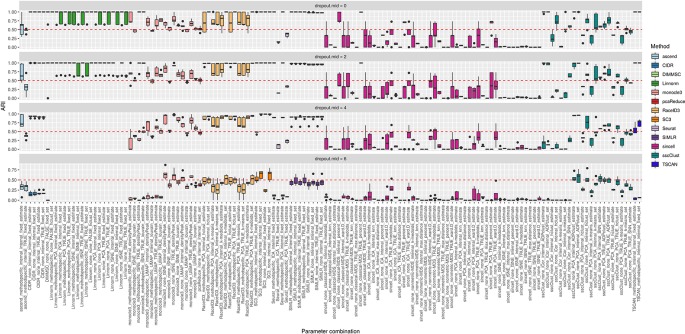
Single-cell RNA-seq (scRNAseq) is a powerful tool to study heterogeneity of cells. Recently, several clustering based methods have been proposed to identify distinct cell populations. These methods are based on different statistical models and usually require to perform several additional steps, such as preprocessing or dimension reduction, before applying the clustering algorithm. Individual steps are often controlled by method-specific parameters, permitting the method to be used in different modes on the same datasets, depending on the user choices. The large number of possibilities that these methods provide can intimidate non-expert users, since the available choices are not always clearly documented. In addition, to date, no large studies have invistigated the role and the impact that these choices can have in different experimental contexts. This work aims to provide new insights into the advantages and drawbacks of scRNAseq clustering methods and describe the ranges of possibilities that are offered to users. In particular, we provide an extensive evaluation of several methods with respect to different modes of usage and parameter settings by applying them to real and simulated datasets that vary in terms of dimensionality, number of cell populations or levels of noise. Remarkably, the results presented here show that great variability in the performance of the models is strongly attributed to the choice of the user-specific parameter settings. We describe several tendencies in the performance attributed to their modes of usage and different types of datasets, and identify which methods are strongly affected by data dimensionality in terms of computational time. Finally, we highlight some open challenges in scRNAseq data clustering, such as those related to the identification of the number of clusters.
Keywords: benchmark; clustering methods; high-dimensional data analysis; parameter sensitivity analysis; single-cell RNA-seq.
Copyright © 2019 Krzak, Raykov, Boukouvalas, Cutillo and Angelini.
Figures

References
-
- Benjamini Y., Hochberg Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B. (Methodological) 57 (1), 289–300. 10.1111/j.2517-6161.1995.tb02031.x - DOI
LinkOut - more resources
Full Text Sources