

Harnessing DNA Double-Strand Break Repair for Cancer Treatment
- PMID: 31921645
- PMCID: PMC6921965
- DOI: 10.3389/fonc.2019.01388
Harnessing DNA Double-Strand Break Repair for Cancer Treatment
Abstract
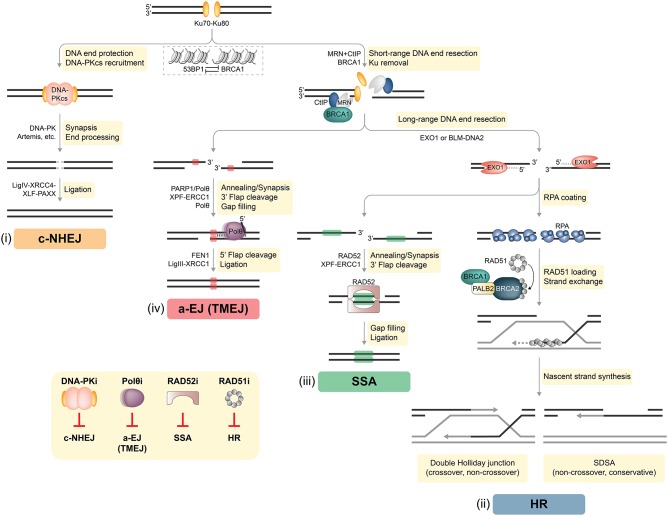
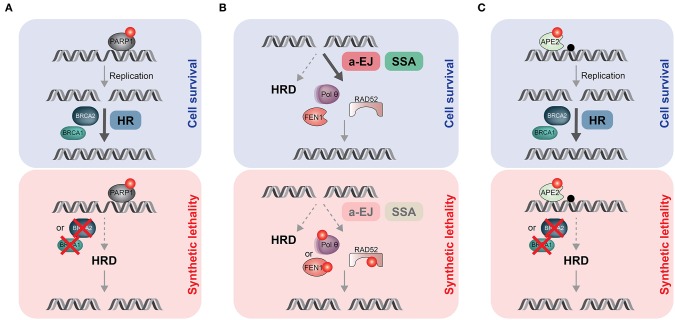
DNA double-strand breaks (DSBs) are highly deleterious, with a single unrepaired DSB being sufficient to trigger cell death. Compared to healthy cells, cancer cells have a higher DSB burden due to oncogene-induced replication stress and acquired defects in DNA damage response (DDR) mechanisms. Consequently, hyperproliferating cancer cells rely on efficient DSB repair for their survival. Moreover, augmented DSB repair capacity is a major cause of radio- and chemoresistance and, ultimately, cancer recurrence. Although inherited DDR defects can predispose individuals to develop certain cancers, the very same vulnerability may be therapeutically exploited to preferentially kill tumor cells. A paradigm for DNA repair targeted therapy has emerged in cancers that exhibit mutations in BRCA1 or BRCA2 tumor suppressor genes, conferring a strong defect in homologous recombination, a major and error-free DSB repair pathway. Clinical validation of such approaches, commonly described as synthetic lethality (SL), has been provided by the regulatory approval of poly(ADP-ribose) polymerase 1 inhibitors (PARPi) as monotherapy for BRCA1/2-mutated breast and ovarian tumors. In this review, we will describe the different DSB repair mechanisms and discuss how their specific features could be exploited for cancer therapy. A major emphasis is put on advances in combinatorial treatment modalities and SL approaches arising from DSB repair pathway interdependencies.
Keywords: BRCA; DNA polymerase theta; DSB repair; PARP inhibition (PARPi); alternative end joining (a-EJ); cancer therapy; homologous recombination (HR); synthetic lethality.
Copyright © 2019 Trenner and Sartori.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous