

Evaluation of Phylogenetic Methods for Inferring the Direction of Human Immunodeficiency Virus (HIV) Transmission: HIV Prevention Trials Network (HPTN) 052
- PMID: 31922537
- PMCID: PMC7823077
- DOI: 10.1093/cid/ciz1247
Evaluation of Phylogenetic Methods for Inferring the Direction of Human Immunodeficiency Virus (HIV) Transmission: HIV Prevention Trials Network (HPTN) 052
Abstract
Background: Phylogenetic analysis can be used to assess human immunodeficiency virus (HIV) transmission in populations. We inferred the direction of HIV transmission using whole-genome HIV sequences from couples with known linked infection and known transmission direction.
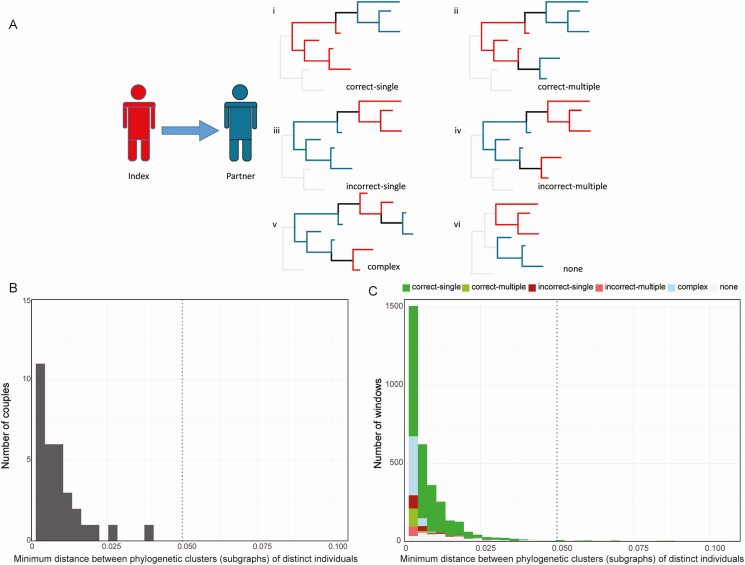
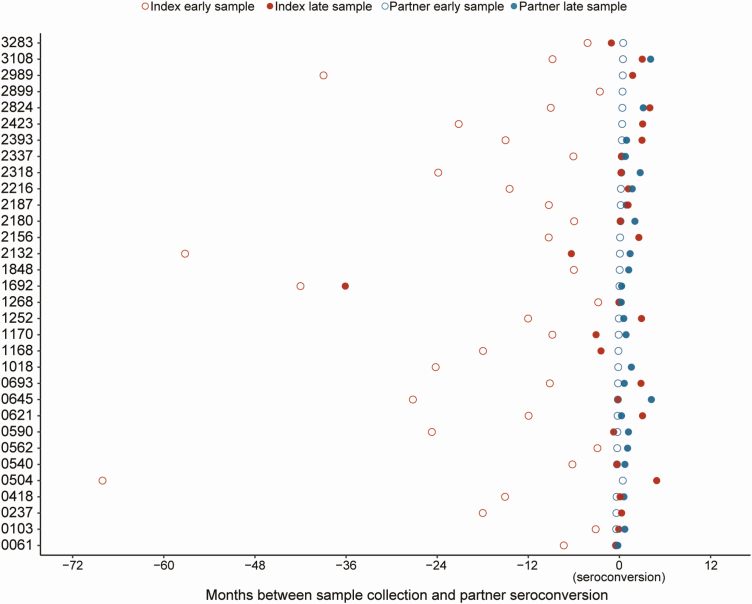
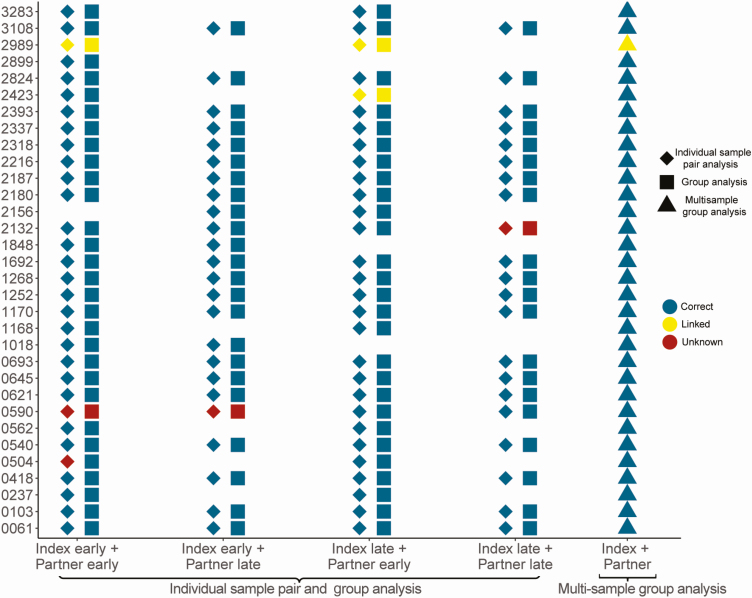
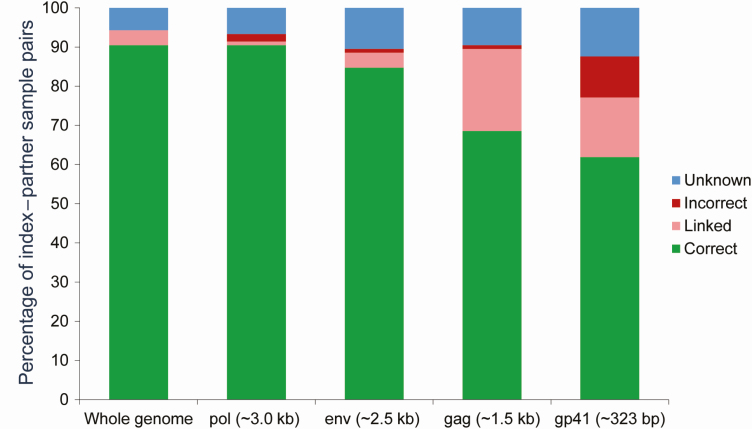
Methods: Complete next-generation sequencing (NGS) data were obtained for 105 unique index-partner sample pairs from 32 couples enrolled in the HIV Prevention Trials Network (HPTN) 052 study (up to 2 samples/person). Index samples were obtained up to 5.5 years before partner infection; partner samples were obtained near the time of seroconversion. The bioinformatics method, phyloscanner, was used to infer transmission direction. Analyses were performed using samples from individual sample pairs, samples from all couples (1 sample/person; group analysis), and all available samples (multisample group analysis). Analysis was also performed using NGS data from defined regions of the HIV genome (gag, pol, env).
Results: Using whole-genome NGS data, transmission direction was inferred correctly (index to partner) for 98 of 105 (93.3%) of the individual sample pairs, 99 of 105 (94.3%) sample pairs using group analysis, and 31 of the 32 couples (96.9%) using multisample group analysis. There were no cases where the incorrect transmission direction (partner to index) was inferred. The accuracy of the method was higher with greater time between index and partner sample collection. Pol region sequences performed better than env or gag sequences for inferring transmission direction.
Conclusions: We demonstrate the potential of a phylogenetic method to infer the direction of HIV transmission between 2 individuals using whole-genome and pol NGS data.
Keywords: HIV; HPTN 052; direction of transmission; next-generation sequencing; phylogenetic analysis.
© The Author(s) 2020. Published by Oxford University Press for the Infectious Diseases Society of America. All rights reserved. For permissions, e-mail: journals.permissions@oup.com.
Figures
Comment in
-
Determining the Direction of Human Immunodeficiency Virus Transmission: Benefits and Potential Harms of Taking Phylogenetic Analysis One Step Further.Clin Infect Dis. 2021 Jan 23;72(1):38-40. doi: 10.1093/cid/ciz1248. Clin Infect Dis. 2021. PMID: 31922539 No abstract available.
References
-
- Hué S, Clewley JP, Cane PA, Pillay D. HIV-1 pol gene variation is sufficient for reconstruction of transmissions in the era of antiretroviral therapy. AIDS 2004; 18:719–28. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
