

Sodium-glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy
- PMID: 31924211
- PMCID: PMC6953156
- DOI: 10.1186/s12933-019-0980-4
Sodium-glucose cotransporter 2 inhibitor Dapagliflozin attenuates diabetic cardiomyopathy
Abstract
Background: Diabetes mellitus type 2 (DM2) is a risk factor for developing heart failure but there is no specific therapy for diabetic heart disease. Sodium glucose transporter 2 inhibitors (SGLT2I) are recently developed diabetic drugs that primarily work on the kidney. Clinical data describing the cardiovascular benefits of SGLT2Is highlight the potential therapeutic benefit of these drugs in the prevention of cardiovascular events and heart failure. However, the underlying mechanism of protection remains unclear. We investigated the effect of Dapagliflozin-SGLT2I, on diabetic cardiomyopathy in a mouse model of DM2.
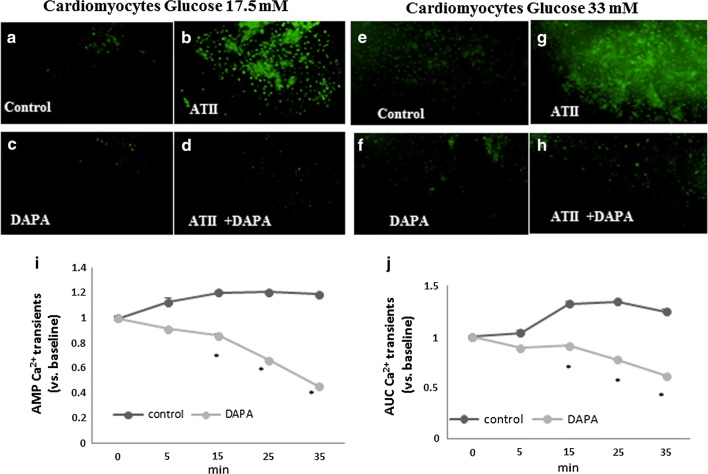
Methods: Cardiomyopathy was induced in diabetic mice (db/db) by subcutaneous infusion of angiotensin II (ATII) for 30 days using an osmotic pump. Dapagliflozin (1.5 mg/kg/day) was administered concomitantly in drinking water. Male homozygous, 12-14 weeks old WT or db/db mice (n = 4-8/group), were used for the experiments. Isolated cardiomyocytes were exposed to glucose (17.5-33 mM) and treated with Dapagliflozin in vitro. Intracellular calcium transients were measured using a fluorescent indicator indo-1.
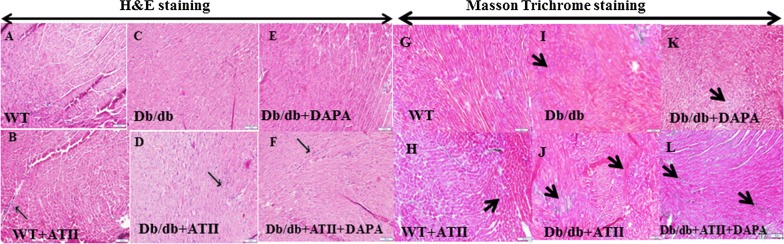
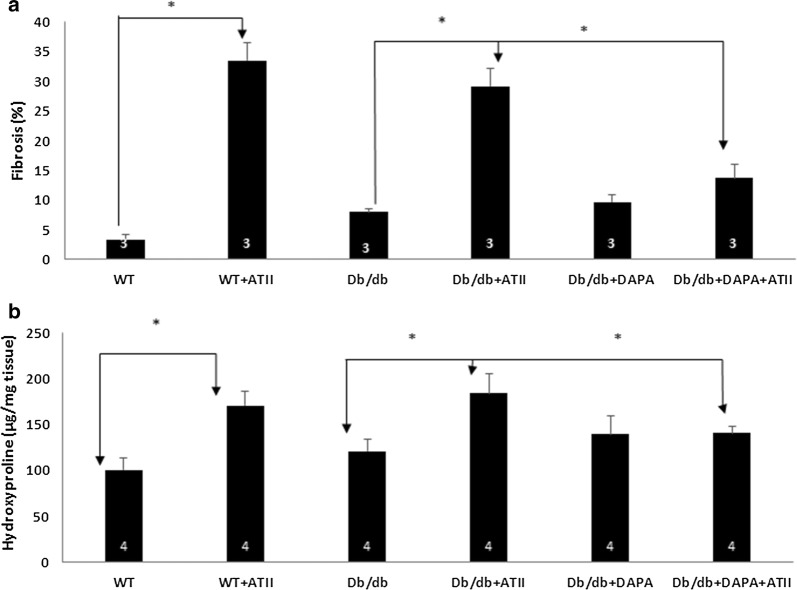
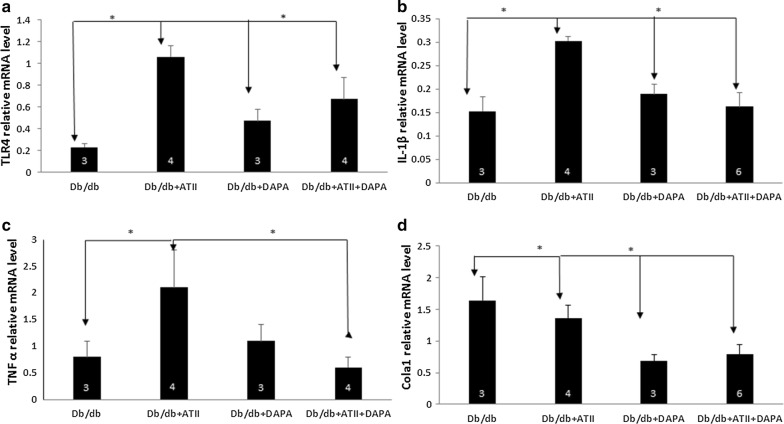
Results: Angiotensin II infusion induced cardiomyopathy in db/db mice, manifested by cardiac hypertrophy, myocardial fibrosis and inflammation (TNFα, TLR4). Dapagliflozin decreased blood glucose (874 ± 111 to 556 ± 57 mg/dl, p < 0.05). In addition it attenuated fibrosis and inflammation and increased the left ventricular fractional shortening in ATII treated db/db mice. In isolated cardiomyocytes Dapagliflozin decreased intracellular calcium transients, inflammation and ROS production. Finally, voltage-dependent L-type calcium channel (CACNA1C), the sodium-calcium exchanger (NCX) and the sodium-hydrogen exchanger 1 (NHE) membrane transporters expression was reduced following Dapagliflozin treatment.
Conclusion: Dapagliflozin was cardioprotective in ATII-stressed diabetic mice. It reduced oxygen radicals, as well the activity of membrane channels related to calcium transport. The cardioprotective effect manifested by decreased fibrosis, reduced inflammation and improved systolic function. The clinical implication of our results suggest a novel pharmacologic approach for the treatment of diabetic cardiomyopathy through modulation of ion homeostasis.
Keywords: Calcium transport fibrosis; Cardiomyocytes; Cardiomyopathy; Dapagliflozin; Diabetes mellitus type 2; Inflammation; ROS.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures


References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
