

Pyruvate Kinase M2 Promotes Expression of Proinflammatory Mediators in House Dust Mite-Induced Allergic Airways Disease
- PMID: 31924651
- PMCID: PMC6994838
- DOI: 10.4049/jimmunol.1901086
Pyruvate Kinase M2 Promotes Expression of Proinflammatory Mediators in House Dust Mite-Induced Allergic Airways Disease
Abstract
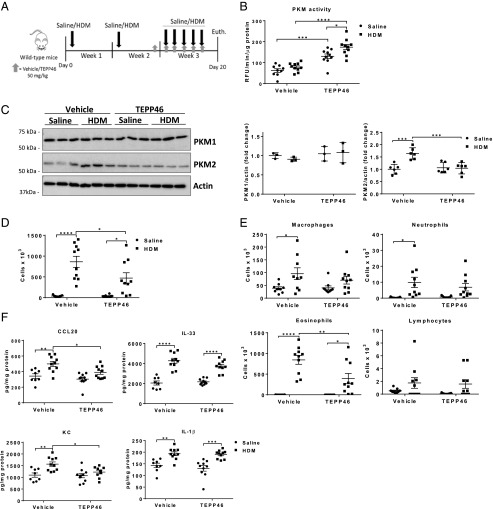
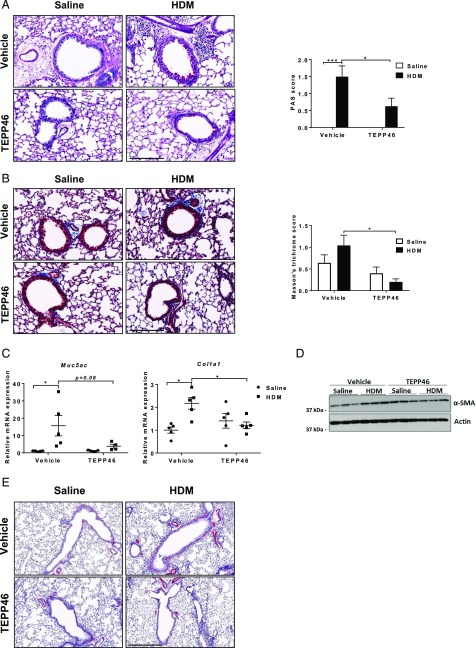
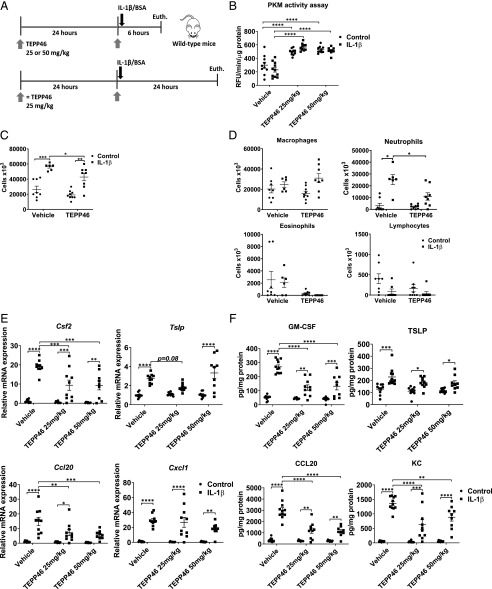
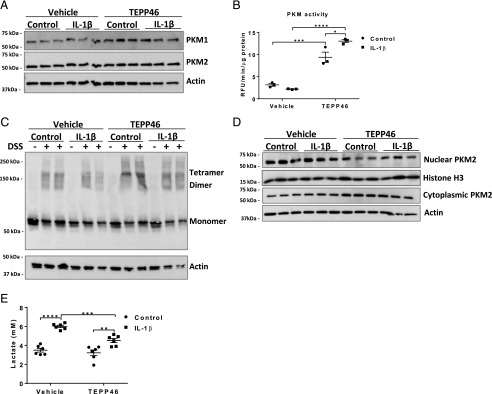
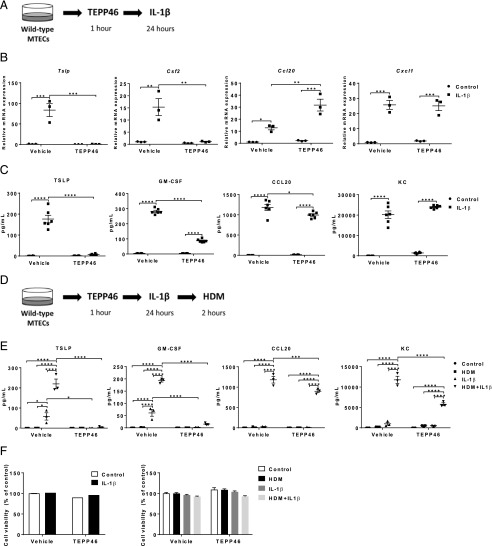
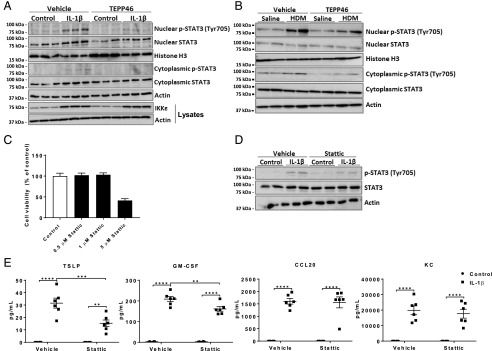
Asthma is a chronic disorder characterized by inflammation, mucus metaplasia, airway remodeling, and hyperresponsiveness. We recently showed that IL-1-induced glycolytic reprogramming contributes to allergic airway disease using a murine house dust mite model. Moreover, levels of pyruvate kinase M2 (PKM2) were increased in this model as well as in nasal epithelial cells from asthmatics as compared with healthy controls. Although the tetramer form of PKM2 converts phosphoenolpyruvate to pyruvate, the dimeric form of PKM2 has alternative, nonglycolysis functions as a transcriptional coactivator to enhance the transcription of several proinflammatory cytokines. In the current study, we examined the impact of PKM2 on the pathogenesis of house dust mite-induced allergic airways disease in C57BL/6NJ mice. We report, in this study, that activation of PKM2, using the small molecule activator, TEPP46, augmented PKM activity in lung tissues and attenuated airway eosinophils, mucus metaplasia, and subepithelial collagen. TEPP46 attenuated IL-1β-mediated airway inflammation and expression of proinflammatory mediators. Exposure to TEPP46 strongly decreased the IL-1β-mediated increases in thymic stromal lymphopoietin (TSLP) and GM-CSF in primary tracheal epithelial cells isolated from C57BL/6NJ mice. We also demonstrate that IL-1β-mediated increases in nuclear phospho-STAT3 were decreased by TEPP46. Finally, STAT3 inhibition attenuated the IL-1β-induced release of TSLP and GM-CSF, suggesting that the ability of PKM2 to phosphorylate STAT3 contributes to its proinflammatory function. Collectively, these results demonstrate that the glycolysis-inactive form of PKM2 plays a crucial role in the pathogenesis of allergic airways disease by increasing IL-1β-induced proinflammatory signaling, in part, through phosphorylation of STAT3.
Copyright © 2020 by The American Association of Immunologists, Inc.
Figures
References
-
- Qian X., Aboushousha R., van de Wetering C., Chia S. B., Amiel E., Schneider R. W., van der Velden J. L. J., Lahue K. G., Hoagland D. A., Casey D. T., et al. 2018. IL-1/inhibitory κB kinase ε-induced glycolysis augment epithelial effector function and promote allergic airways disease. J. Allergy Clin. Immunol. 142: 435–450.e10. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
