

Cold-induced urticarial autoinflammatory syndrome related to factor XII activation
- PMID: 31924766
- PMCID: PMC6954242
- DOI: 10.1038/s41467-019-13984-8
Cold-induced urticarial autoinflammatory syndrome related to factor XII activation
Abstract
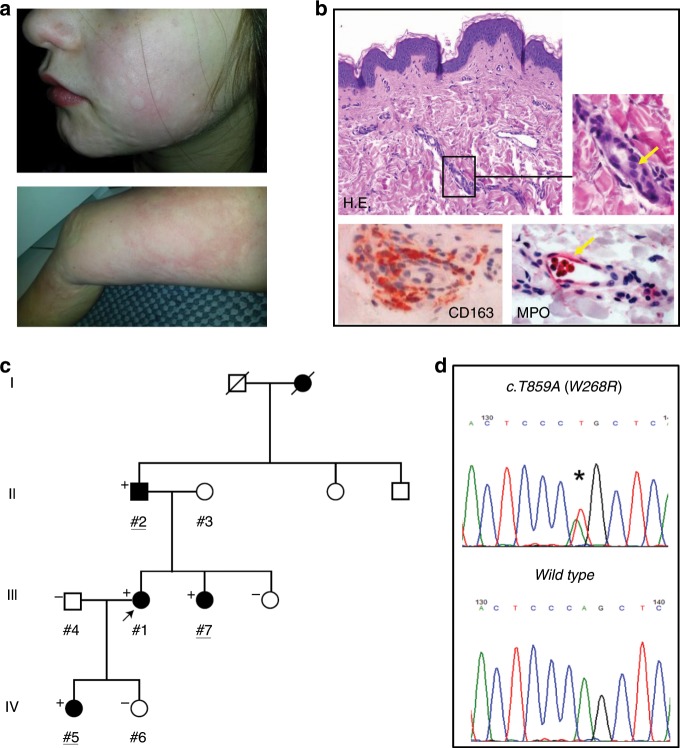
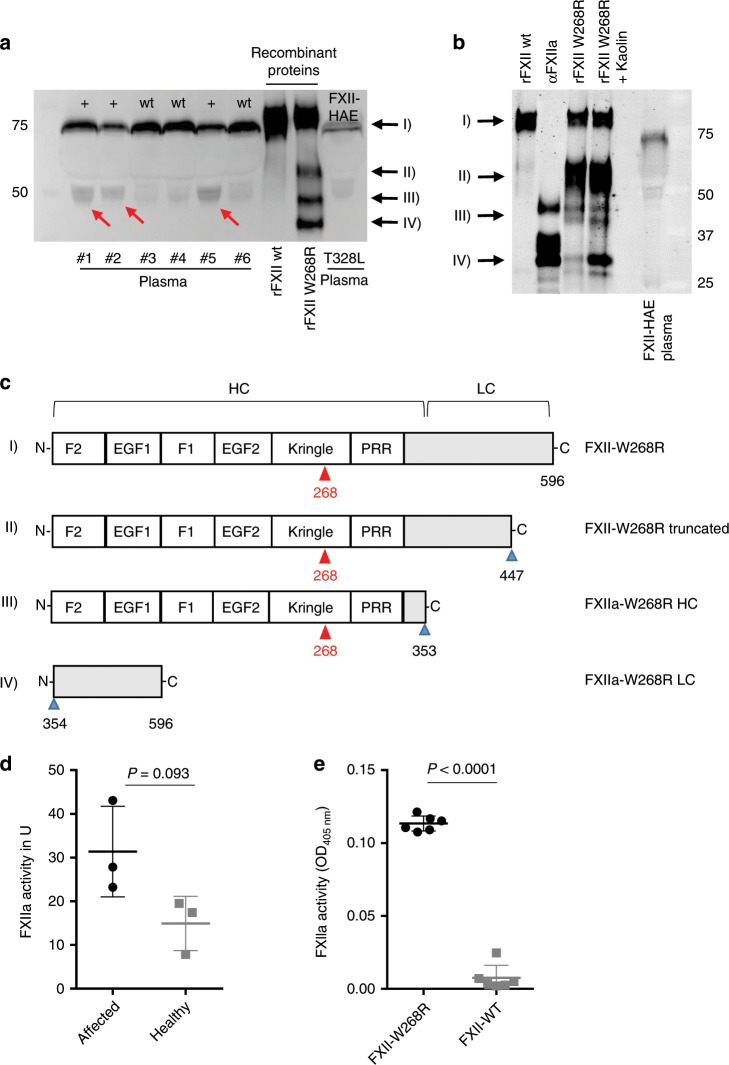
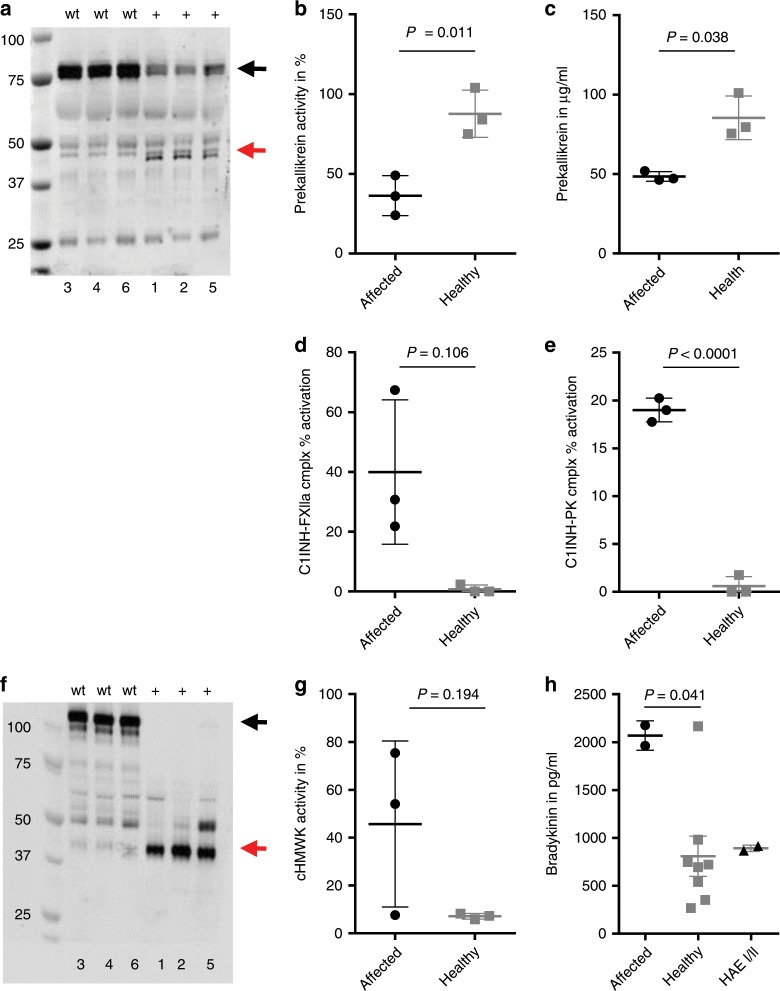
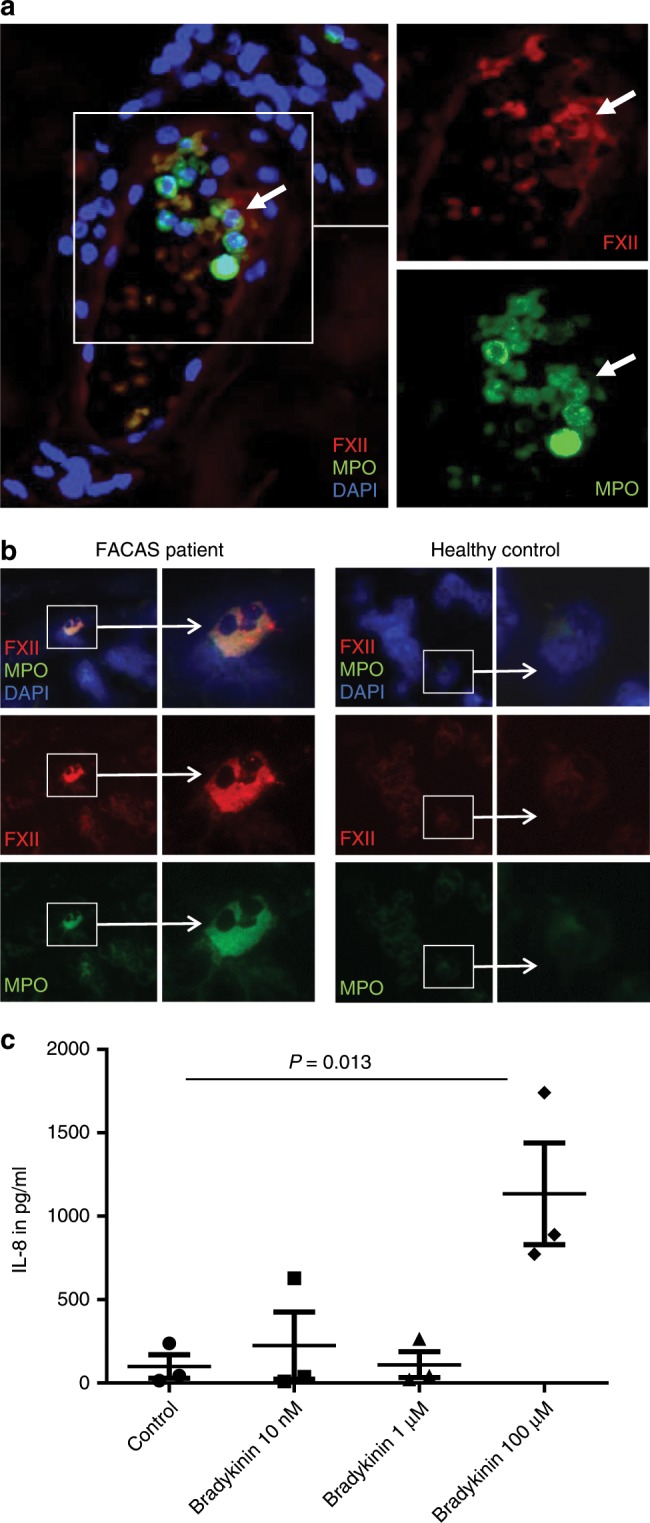
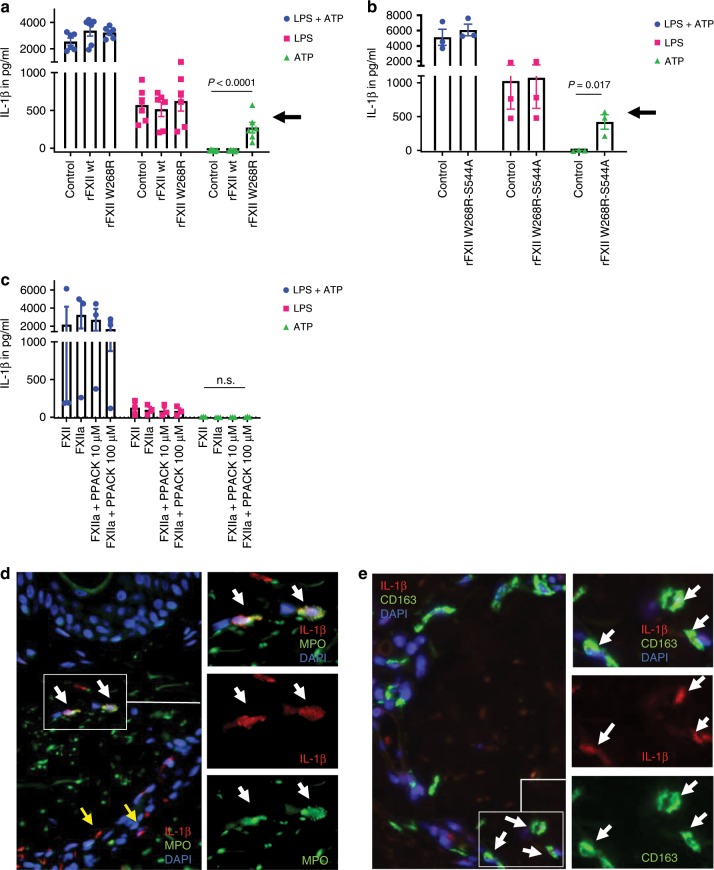
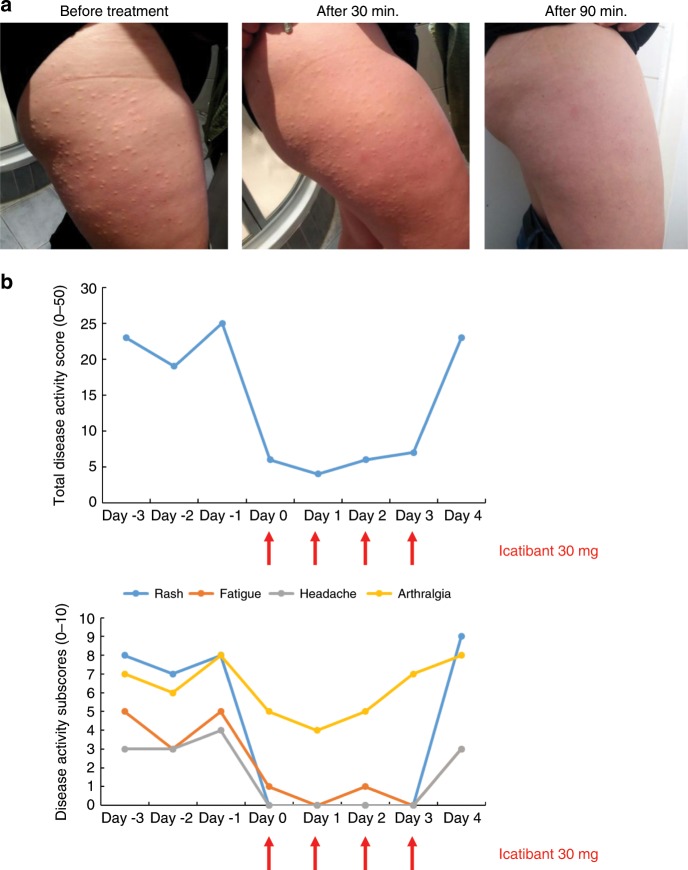
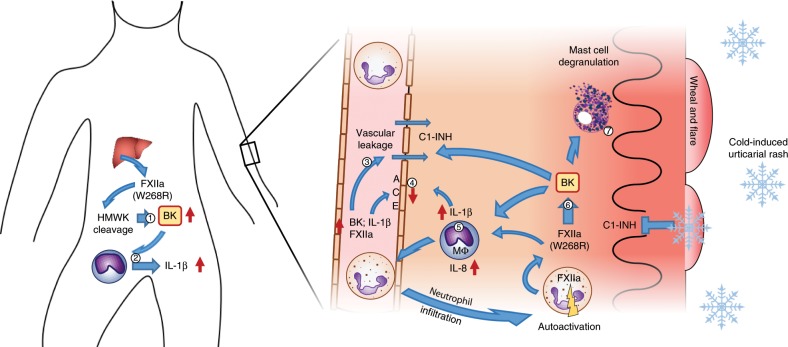
Hereditary autoinflammatory diseases are caused by gene mutations of the innate immune pathway, e.g. nucleotide receptor protein 3 (NLRP3). Here, we report a four-generation family with cold-induced urticarial rash, arthralgia, chills, headache and malaise associated with an autosomal-dominant inheritance. Genetic studies identify a substitution mutation in gene F12 (T859A, resulting in p.W268R) which encodes coagulation factor XII (FXII). Functional analysis reveals enhanced autocatalytic cleavage of the mutated protein and spontaneous FXII activation in patient plasma and in supernatant of transfected HEK293 cells expressing recombinant W268R-mutated proteins. Furthermore, we observe reduced plasma prekallikrein, cleaved high molecular weight kininogen and elevated plasma bradykinin. Neutrophils are identified as a local source of FXII. Interleukin-1β (IL-1β) is upregulated in lesional skin and mononuclear donor cells exposed to recombinant mutant proteins. Treatment with icatibant (bradykinin-B2-antagonist) or anakinra (interleukin-1-antagonist) reduces disease activity in patients. In conclusion, our findings provide a link between contact system activation and cytokine-mediated inflammation.
Conflict of interest statement
M.Mau. received consultation fees and research support from Shire/Takeda outside the submitted work. K.K. received research support from Shire/Takeda outside the submitted work. The remaining authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
