

Artificial escape from XCI by DNA methylation editing of the CDKL5 gene
- PMID: 31925439
- PMCID: PMC7049732
- DOI: 10.1093/nar/gkz1214
Artificial escape from XCI by DNA methylation editing of the CDKL5 gene
Abstract
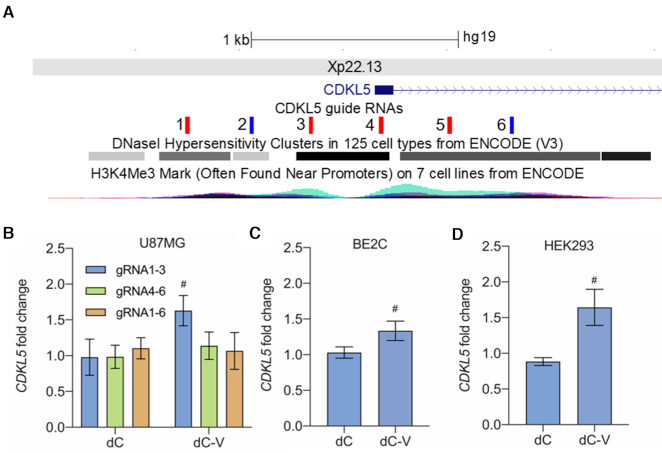
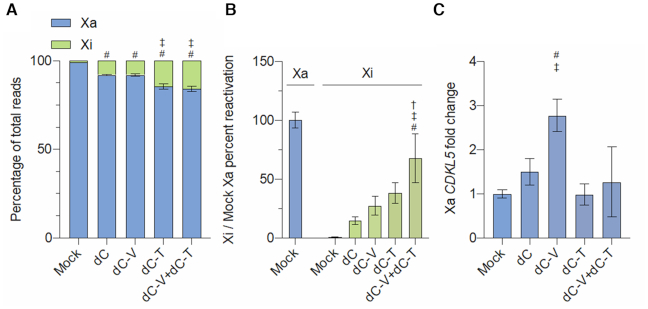
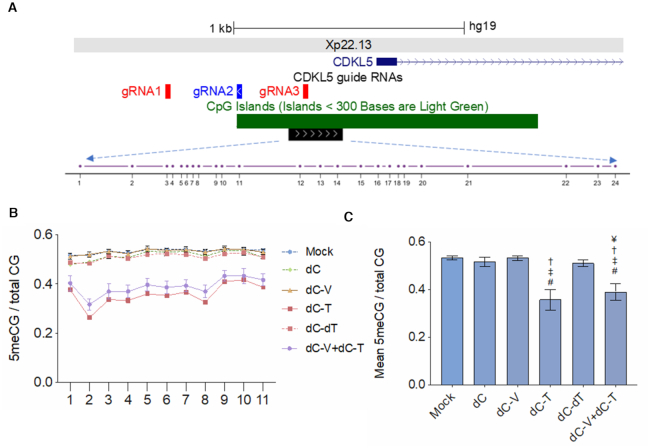
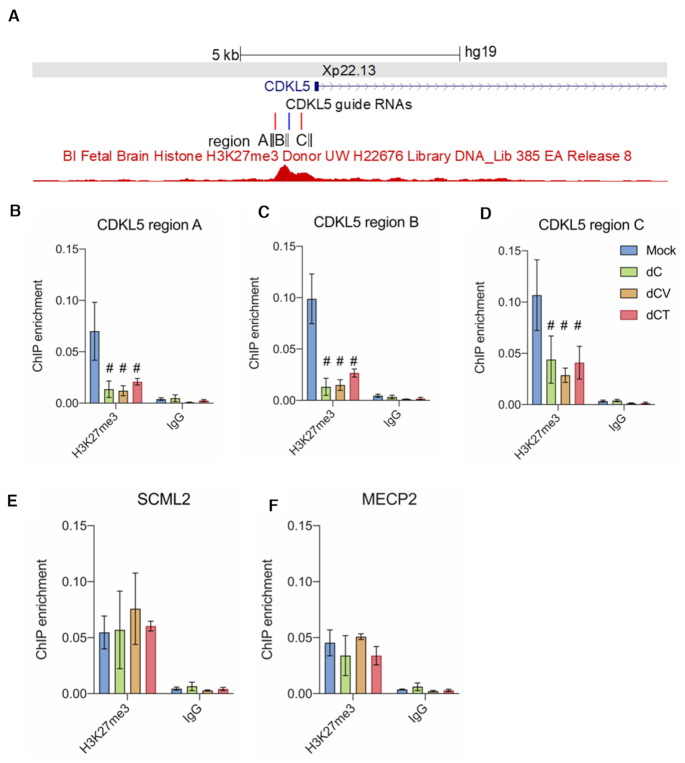
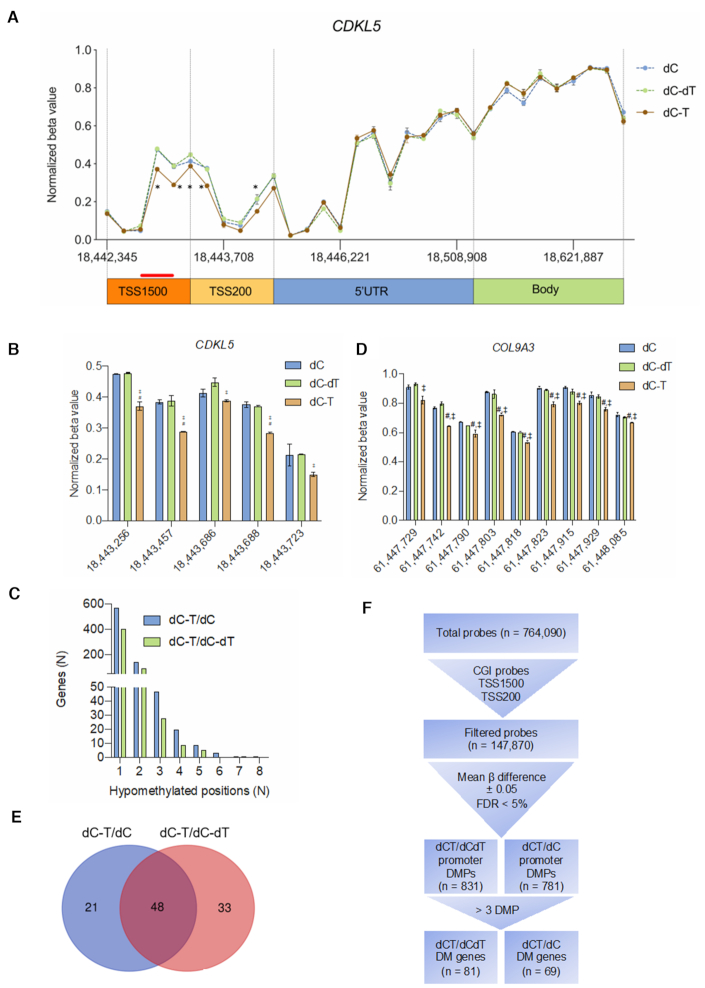
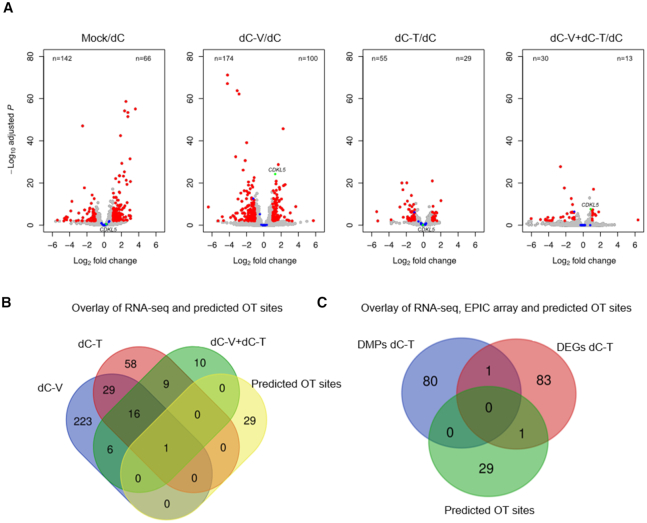
A significant number of X-linked genes escape from X chromosome inactivation and are associated with a distinct epigenetic signature. One epigenetic modification that strongly correlates with X-escape is reduced DNA methylation in promoter regions. Here, we created an artificial escape by editing DNA methylation on the promoter of CDKL5, a gene causative for an infantile epilepsy, from the silenced X-chromosomal allele in human neuronal-like cells. We identify that a fusion of the catalytic domain of TET1 to dCas9 targeted to the CDKL5 promoter using three guide RNAs causes significant reactivation of the inactive allele in combination with removal of methyl groups from CpG dinucleotides. Strikingly, we demonstrate that co-expression of TET1 and a VP64 transactivator have a synergistic effect on the reactivation of the inactive allele to levels >60% of the active allele. We further used a multi-omics assessment to determine potential off-targets on the transcriptome and methylome. We find that synergistic delivery of dCas9 effectors is highly selective for the target site. Our findings further elucidate a causal role for reduced DNA methylation associated with escape from X chromosome inactivation. Understanding the epigenetics associated with escape from X chromosome inactivation has potential for those suffering from X-linked disorders.
© The Author(s) 2020. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
