

Guidelines for cell-type heterogeneity quantification based on a comparative analysis of reference-free DNA methylation deconvolution software
- PMID: 31931698
- PMCID: PMC6958785
- DOI: 10.1186/s12859-019-3307-2
Guidelines for cell-type heterogeneity quantification based on a comparative analysis of reference-free DNA methylation deconvolution software
Abstract
Background: Cell-type heterogeneity of tumors is a key factor in tumor progression and response to chemotherapy. Tumor cell-type heterogeneity, defined as the proportion of the various cell-types in a tumor, can be inferred from DNA methylation of surgical specimens. However, confounding factors known to associate with methylation values, such as age and sex, complicate accurate inference of cell-type proportions. While reference-free algorithms have been developed to infer cell-type proportions from DNA methylation, a comparative evaluation of the performance of these methods is still lacking.
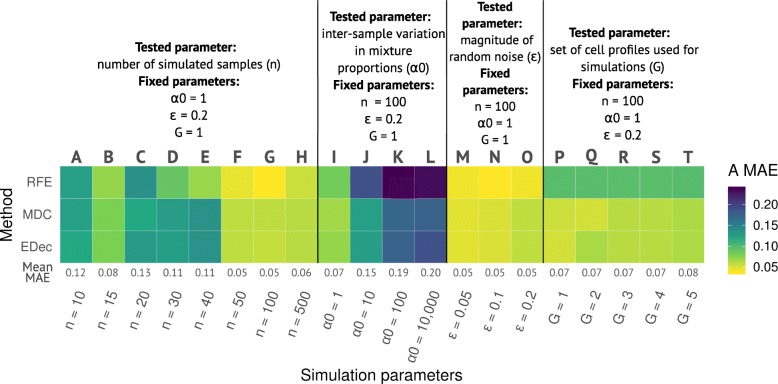
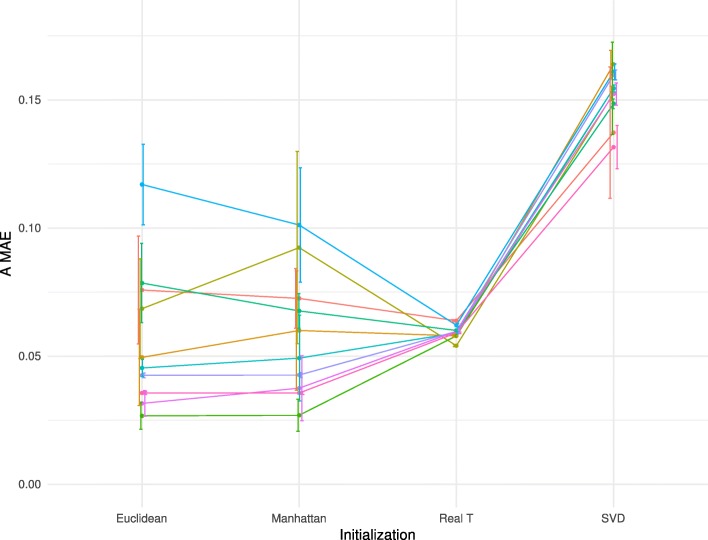
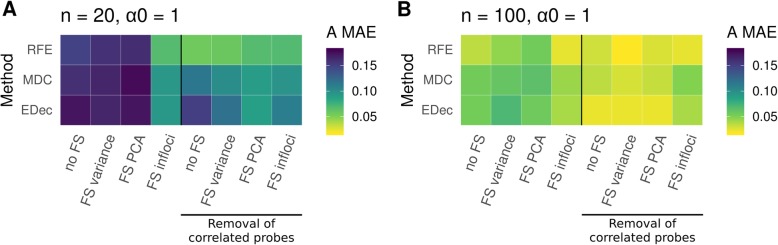
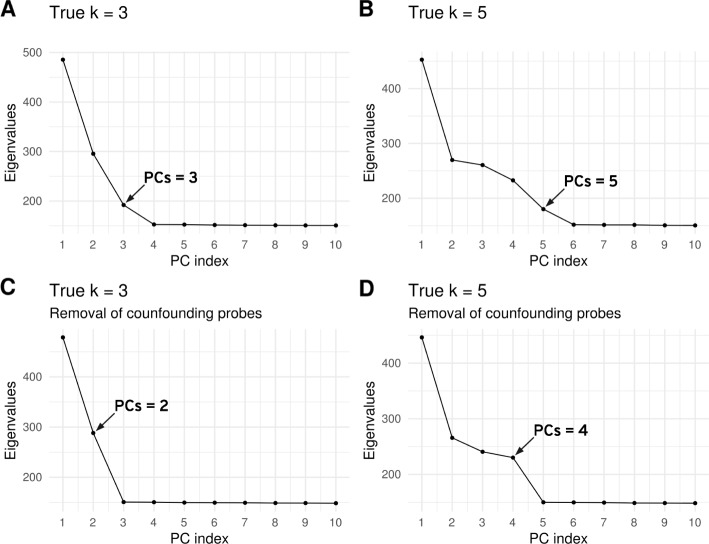
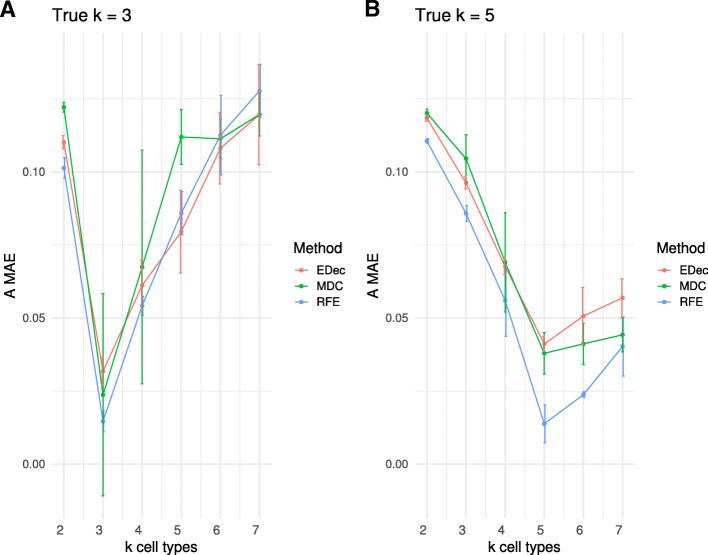
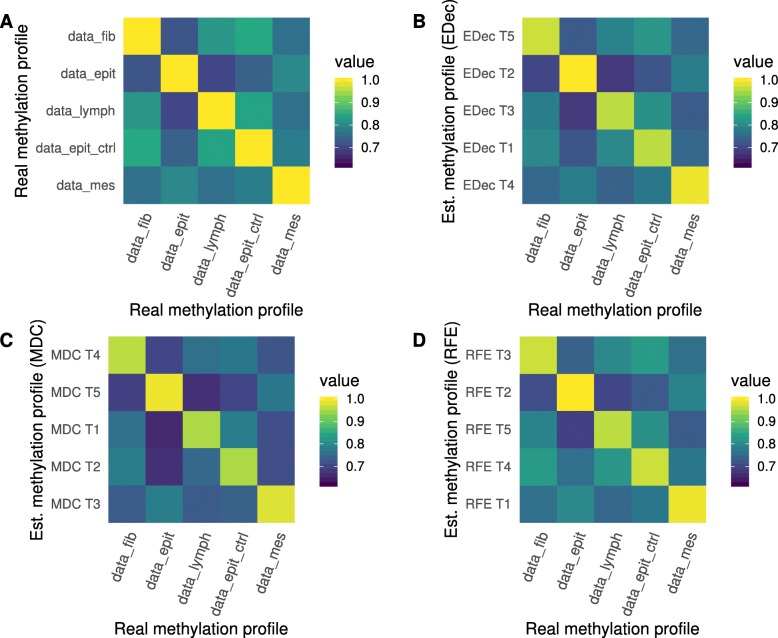
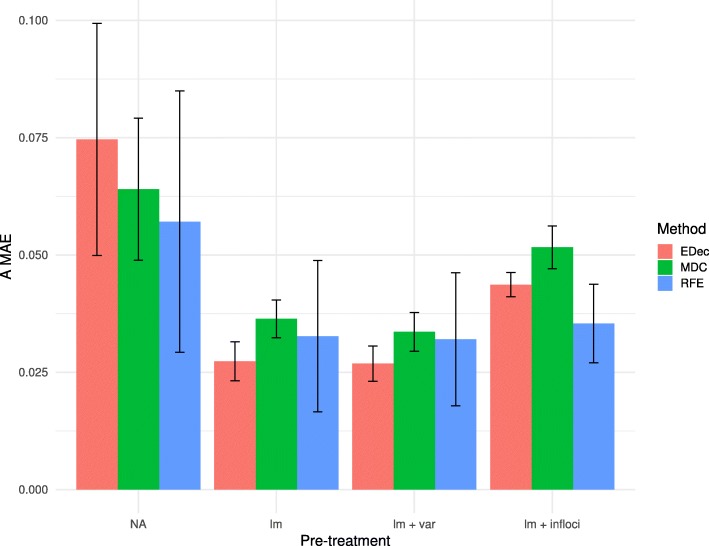
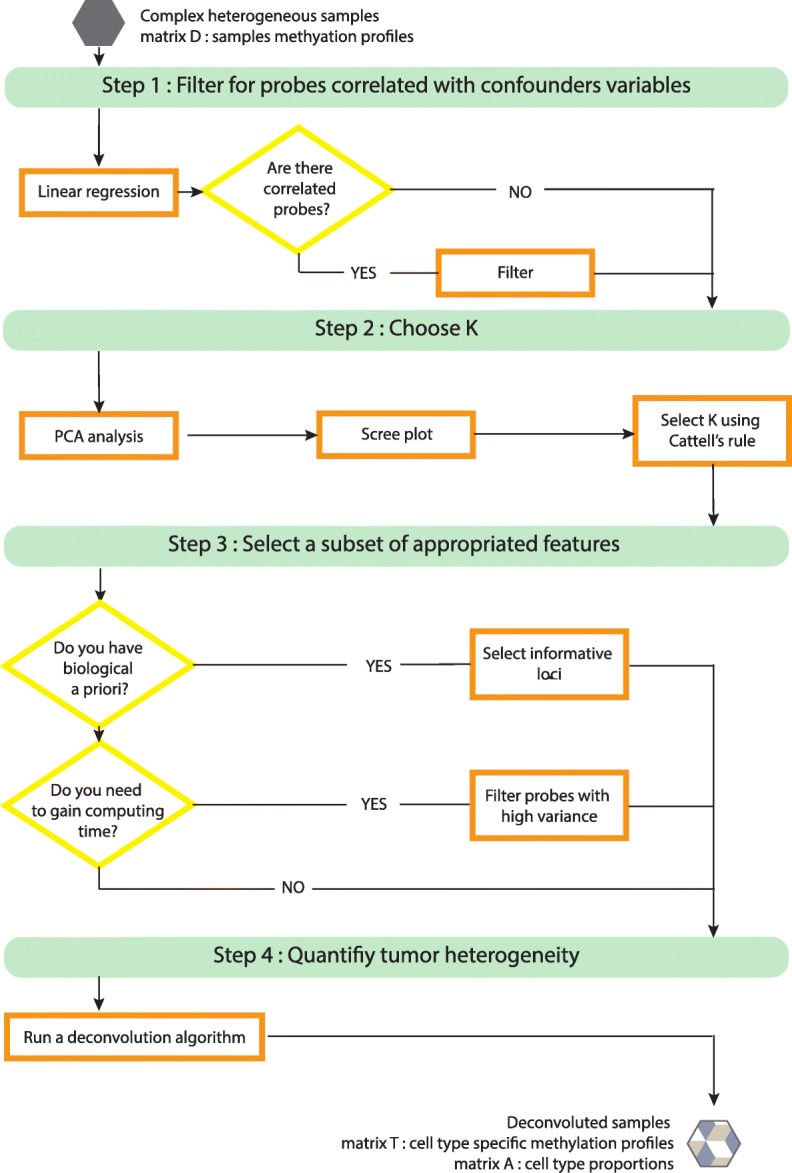
Results: Here we use simulations to evaluate several computational pipelines based on the software packages MeDeCom, EDec, and RefFreeEWAS. We identify that accounting for confounders, feature selection, and the choice of the number of estimated cell types are critical steps for inferring cell-type proportions. We find that removal of methylation probes which are correlated with confounder variables reduces the error of inference by 30-35%, and that selection of cell-type informative probes has similar effect. We show that Cattell's rule based on the scree plot is a powerful tool to determine the number of cell-types. Once the pre-processing steps are achieved, the three deconvolution methods provide comparable results. We observe that all the algorithms' performance improves when inter-sample variation of cell-type proportions is large or when the number of available samples is large. We find that under specific circumstances the methods are sensitive to the initialization method, suggesting that averaging different solutions or optimizing initialization is an avenue for future research.
Conclusion: Based on the lessons learned, to facilitate pipeline validation and catalyze further pipeline improvement by the community, we develop a benchmark pipeline for inference of cell-type proportions and implement it in the R package medepir.
Keywords: Cell heterogeneity; DNA methylation; Deconvolution; Epigenetics; Matrix factorization; R package/pipeline.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
