

Assessment of cellular cobalamin metabolism in Gaucher disease
- PMID: 31931749
- PMCID: PMC6958775
- DOI: 10.1186/s12881-020-0947-z
Assessment of cellular cobalamin metabolism in Gaucher disease
Abstract
Background: Gaucher disease (GD) is a lysosomal disorder caused by biallelic pathogenic mutations in the GBA1 gene that encodes beta-glucosidase (GCase), and more rarely, by a deficiency in the GCase activator, saposin C. Clinically, GD manifests with heterogeneous multiorgan involvement mainly affecting hematological, hepatic and neurological axes. This disorder is divided into three types, based on the absence (type I) or presence and severity (types II and III) of involvement of the central nervous system. At the cellular level, deficiency of GBA1 disturbs lysosomal storage with buildup of glucocerebroside. The consequences of disturbed lysosomal metabolism on biochemical pathways that require lysosomal processing are unknown. Abnormal systemic markers of cobalamin (Cbl, B12) metabolism have been reported in patients with GD, suggesting impairments in lysosomal handling of Cbl or in its downstream utilization events.
Methods: Cultured skin fibroblasts from control humans (n = 3), from patients with GD types I (n = 1), II (n = 1) and III (n = 1) and an asymptomatic carrier of GD were examined for their GCase enzymatic activity and lysosomal compartment intactness. Control human and GD fibroblasts were cultured in growth medium with and without 500 nM hydroxocobalamin supplementation. Cellular cobalamin status was examined via determination of metabolomic markers in cell lysate (intracellular) and conditioned culture medium (extracellular). The presence of transcobalamin (TC) in whole cell lysates was examined by Western blot.
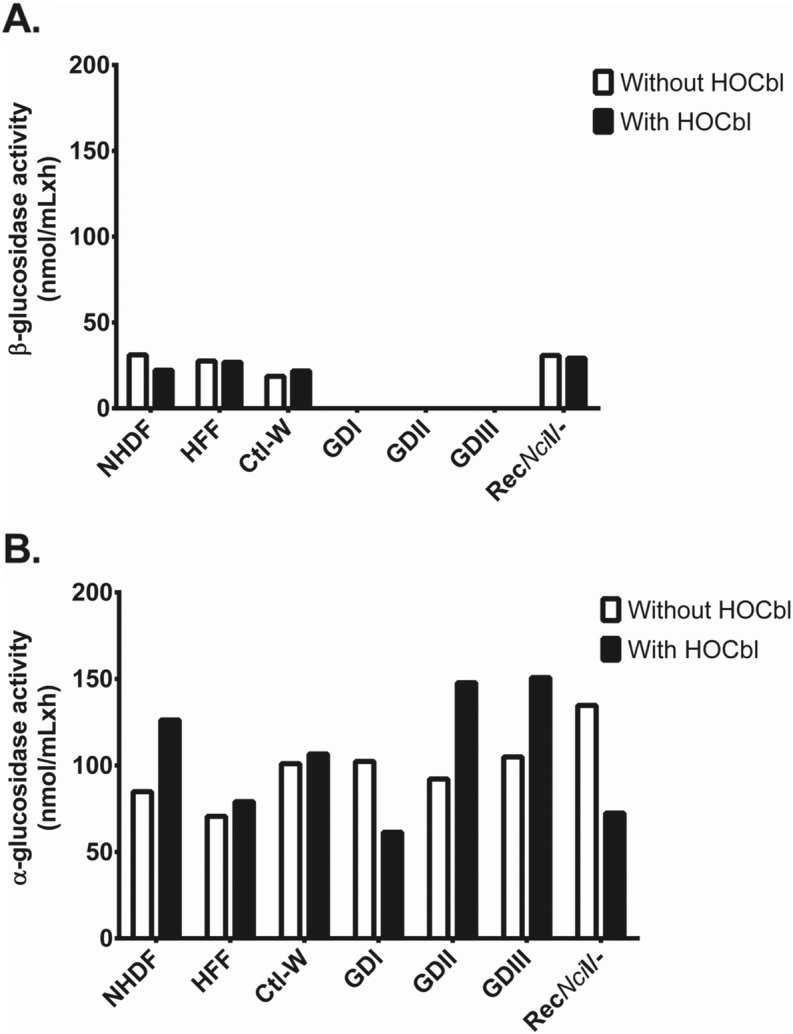
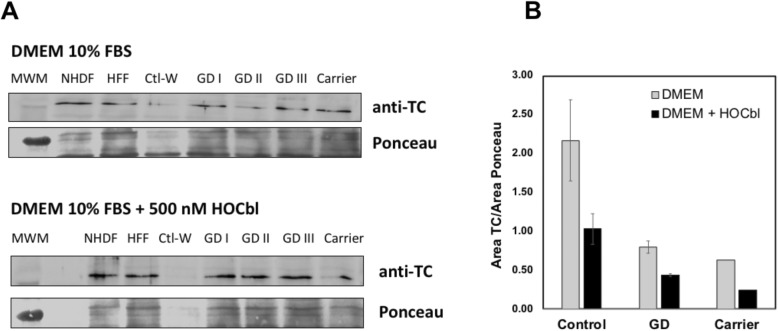
Results: Cultured skin fibroblasts from GD patients exhibited reduced GCase activity compared to healthy individuals and an asymptomatic carrier of GD, demonstrating a preserved disease phenotype in this cell type. The concentrations of total homocysteine (tHcy), methylmalonic acid (MMA), cysteine (Cys) and methionine (Met) in GD cells were comparable to control levels, except in one patient with GD III. The response of these metabolomic markers to supplementation with hydroxocobalamin (HOCbl) yielded variable results. The content of transcobalamin in whole cell lysates was comparable in control human and GD patients.
Conclusions: Our results indicate that cobalamin transport and cellular processing pathways are overall protected from lysosomal storage damage in GD fibroblasts. Extending these studies to hepatocytes, macrophages and plasma will shed light on cell- and compartment-specific vitamin B12 metabolism in Gaucher disease.
Keywords: Beta-glucosidase; Cobalamin; Gaucher disease; Homocysteine; Methylmalonic acid; Transcobalamin; Vitamin B12.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures
Similar articles
-
Deficiency of Glucocerebrosidase Activity beyond Gaucher Disease: PSAP and LIMP-2 Dysfunctions.Int J Mol Sci. 2024 Jun 16;25(12):6615. doi: 10.3390/ijms25126615. Int J Mol Sci. 2024. PMID: 38928321 Free PMC article.
-
Saposin C mutations in Gaucher disease patients resulting in lysosomal lipid accumulation, saposin C deficiency, but normal prosaposin processing and sorting.Hum Mol Genet. 2010 Aug 1;19(15):2987-97. doi: 10.1093/hmg/ddq204. Epub 2010 May 19. Hum Mol Genet. 2010. PMID: 20484222
-
Elevated holo-transcobalamin in Gaucher disease type II: A case report.Am J Med Genet A. 2021 Aug;185(8):2471-2476. doi: 10.1002/ajmg.a.62252. Epub 2021 May 24. Am J Med Genet A. 2021. PMID: 34031990 Review.
-
Lysosomal functions and dysfunctions: Molecular and cellular mechanisms underlying Gaucher disease and its association with Parkinson disease.Adv Drug Deliv Rev. 2022 Aug;187:114402. doi: 10.1016/j.addr.2022.114402. Epub 2022 Jun 25. Adv Drug Deliv Rev. 2022. PMID: 35764179 Review.
-
Gaucher disease iPSC-derived osteoblasts have developmental and lysosomal defects that impair bone matrix deposition.Hum Mol Genet. 2018 Mar 1;27(5):811-822. doi: 10.1093/hmg/ddx442. Hum Mol Genet. 2018. PMID: 29301038 Free PMC article.
References
-
- Beutler E, Grabowski GA, et al. Gaucher disease. In: Scriver CBA, Beaudet AL, et al., editors. The metabolic & molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3635–3668.
-
- Grabowski GA, Petsko GA, Kolodny EH, et al. Gaucher Disease. In: Valle D, Beaudet AL, et al., editors. OMMBID – the Online Metabolic & Molecular Bases of inherited disease. New York: McGraw-Hill; 2013.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
