

Distinct DNA repair pathways cause genomic instability at alternative DNA structures
- PMID: 31932649
- PMCID: PMC6957503
- DOI: 10.1038/s41467-019-13878-9
Distinct DNA repair pathways cause genomic instability at alternative DNA structures
Abstract
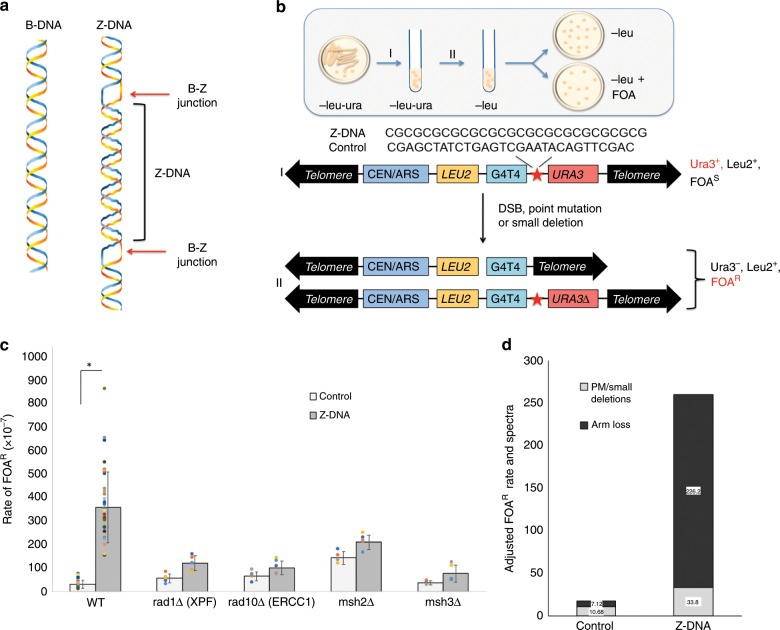
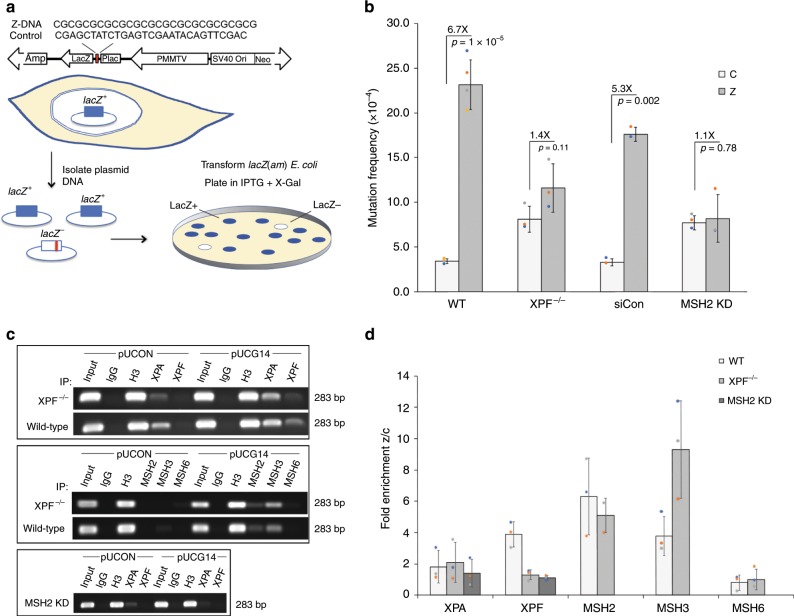
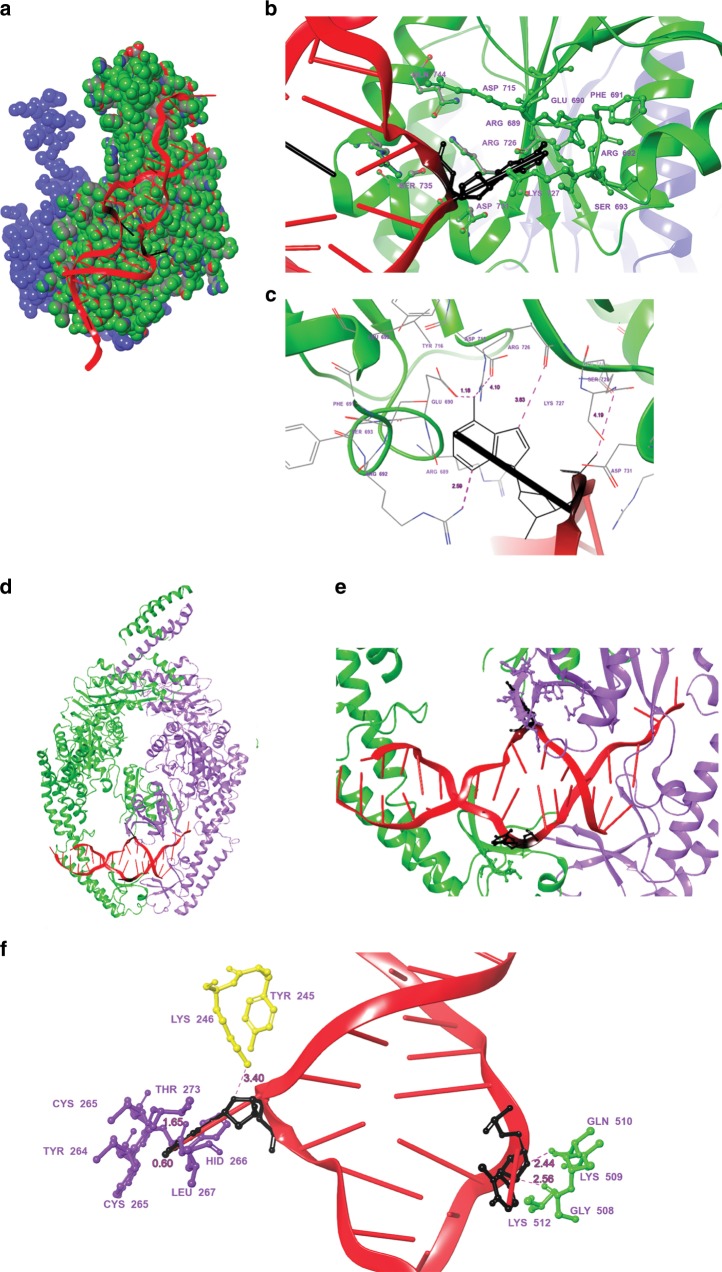
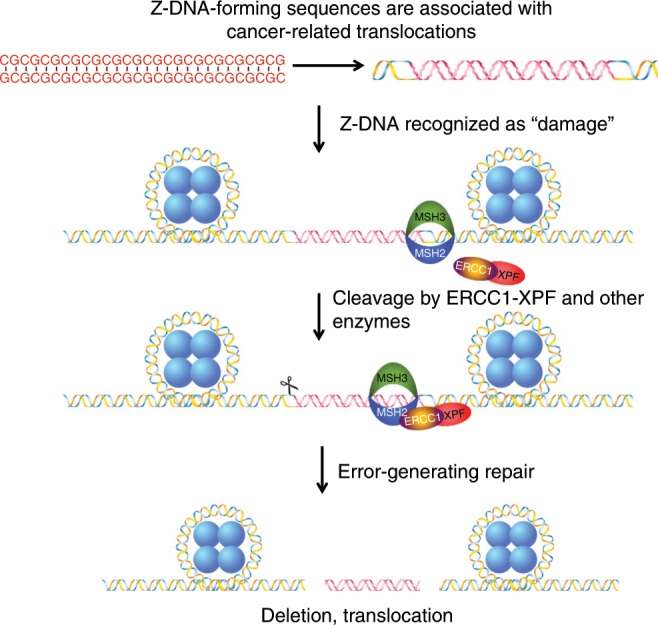
Alternative DNA structure-forming sequences can stimulate mutagenesis and are enriched at mutation hotspots in human cancer genomes, implicating them in disease etiology. However, the mechanisms involved are not well characterized. Here, we discover that Z-DNA is mutagenic in yeast as well as human cells, and that the nucleotide excision repair complex, Rad10-Rad1(ERCC1-XPF), and the mismatch repair complex, Msh2-Msh3, are required for Z-DNA-induced genetic instability in yeast and human cells. Both ERCC1-XPF and MSH2-MSH3 bind to Z-DNA-forming sequences, though ERCC1-XPF recruitment to Z-DNA is dependent on MSH2-MSH3. Moreover, ERCC1-XPF-dependent DNA strand-breaks occur near the Z-DNA-forming region in human cell extracts, and we model these interactions at the sub-molecular level. We propose a relationship in which these complexes recognize and process Z-DNA in eukaryotes, representing a mechanism of Z-DNA-induced genomic instability.
Conflict of interest statement
The authors declare no competing interests.
Figures

Similar articles
-
Distinct roles of XPF-ERCC1 and Rad1-Rad10-Saw1 in replication-coupled and uncoupled inter-strand crosslink repair.Nat Commun. 2018 May 23;9(1):2025. doi: 10.1038/s41467-018-04327-0. Nat Commun. 2018. PMID: 29795289 Free PMC article.
-
Coordination of Rad1-Rad10 interactions with Msh2-Msh3, Saw1 and RPA is essential for functional 3' non-homologous tail removal.Nucleic Acids Res. 2018 Jun 1;46(10):5075-5096. doi: 10.1093/nar/gky254. Nucleic Acids Res. 2018. PMID: 29660012 Free PMC article.
-
Requirement of yeast Rad1-Rad10 nuclease for the removal of 3'-blocked termini from DNA strand breaks induced by reactive oxygen species.Genes Dev. 2004 Sep 15;18(18):2283-91. doi: 10.1101/gad.1232804. Epub 2004 Sep 1. Genes Dev. 2004. PMID: 15371342 Free PMC article.
-
The ERCC1 and ERCC4 (XPF) genes and gene products.Gene. 2015 Sep 15;569(2):153-61. doi: 10.1016/j.gene.2015.06.026. Epub 2015 Jun 12. Gene. 2015. PMID: 26074087 Free PMC article. Review.
-
Alkylation damage in DNA and RNA--repair mechanisms and medical significance.DNA Repair (Amst). 2004 Nov 2;3(11):1389-407. doi: 10.1016/j.dnarep.2004.05.004. DNA Repair (Amst). 2004. PMID: 15380096 Review.
Cited by
-
Gerontology through the Eyes of 21st Century Toxicology.Chem Res Toxicol. 2022 Mar 21;35(3):337-339. doi: 10.1021/acs.chemrestox.1c00336. Epub 2022 Jan 5. Chem Res Toxicol. 2022. PMID: 34985863 Free PMC article.
-
Special Issue: A, B and Z: The Structure, Function and Genetics of Z-DNA and Z-RNA.Int J Mol Sci. 2021 Jul 19;22(14):7686. doi: 10.3390/ijms22147686. Int J Mol Sci. 2021. PMID: 34299306 Free PMC article.
-
HLTF resolves G4s and promotes G4-induced replication fork slowing to maintain genome stability.Mol Cell. 2024 Aug 22;84(16):3044-3060.e11. doi: 10.1016/j.molcel.2024.07.018. Epub 2024 Aug 13. Mol Cell. 2024. PMID: 39142279 Free PMC article.
-
The nuclease activity of DNA2 promotes exonuclease 1-independent mismatch repair.J Biol Chem. 2022 Apr;298(4):101831. doi: 10.1016/j.jbc.2022.101831. Epub 2022 Mar 15. J Biol Chem. 2022. PMID: 35300981 Free PMC article.
-
Z-DNA and Z-RNA: Methods-Past and Future.Methods Mol Biol. 2023;2651:295-329. doi: 10.1007/978-1-0716-3084-6_21. Methods Mol Biol. 2023. PMID: 36892776
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
